



Jacob Hepkema
@jhepkema.bsky.social
Postdoc in Lars Velten's lab @crg.eu, working on gene regulation in haematopoiesis using ML techniques
https://jacobhepkema.github.io
https://jacobhepkema.github.io
Reposted by Jacob Hepkema

We are looking for a postdoc to join our team! If you're interested in translating a cutting edge genomics technology (www.nature.com/articles/s41...) to real-life applications in hematology, this is for you. We offer a unique working environment ON THE BEACH: recruitment.crg.eu/content/jobs...

November 20, 2025 at 9:36 AM
We are looking for a postdoc to join our team! If you're interested in translating a cutting edge genomics technology (www.nature.com/articles/s41...) to real-life applications in hematology, this is for you. We offer a unique working environment ON THE BEACH: recruitment.crg.eu/content/jobs...
Reposted by Jacob Hepkema

New 🧬✂️ pre-print! We show that paired prime editing can efficiently generate large deletions — even >1 Mb — with high precision and at scale. We use this to perform the first pooled prime deletion screen across the human genome.
🔗 biorxiv.org/content/10.1...
A short thread (by Juliane Weller)👇
🔗 biorxiv.org/content/10.1...
A short thread (by Juliane Weller)👇

Generating long deletions across the genome with pooled paired prime editing screens
Engineered deletions are a powerful probe for studying genome architecture, function, and regulation. Yet, the lack of effective methods to create them in large numbers and at multi-kilobase scale has...
biorxiv.org
November 5, 2025 at 2:17 PM
New 🧬✂️ pre-print! We show that paired prime editing can efficiently generate large deletions — even >1 Mb — with high precision and at scale. We use this to perform the first pooled prime deletion screen across the human genome.
🔗 biorxiv.org/content/10.1...
A short thread (by Juliane Weller)👇
🔗 biorxiv.org/content/10.1...
A short thread (by Juliane Weller)👇
Reposted by Jacob Hepkema

TF-MAPS: fast high-resolution functional and allosteric mapping of DNA-binding proteins by @XianghuaLi2
Are Transcription Factors really 'undruggable'?
www.biorxiv.org/content/10.1...
Are Transcription Factors really 'undruggable'?
www.biorxiv.org/content/10.1...

TF-MAPS: fast high-resolution functional and allosteric mapping of DNA-binding proteins
Transcription factors (TFs) bind specific DNA sequences to control gene expression. Modulating TF activity is of considerable therapeutic interest but very few TFs have been successfully drugged. TF D...
www.biorxiv.org
October 23, 2025 at 12:50 PM
TF-MAPS: fast high-resolution functional and allosteric mapping of DNA-binding proteins by @XianghuaLi2
Are Transcription Factors really 'undruggable'?
www.biorxiv.org/content/10.1...
Are Transcription Factors really 'undruggable'?
www.biorxiv.org/content/10.1...
Reposted by Jacob Hepkema

Excited / nervous to share the “magnum opus” of my postdoc in Andreas Wagner’s lab!
"De-novo promoters emerge more readily from random DNA than from genomic DNA"
This project is the accumulation of 4 years of work, and lays the foundation for my future group. In short, we… (1/4)
"De-novo promoters emerge more readily from random DNA than from genomic DNA"
This project is the accumulation of 4 years of work, and lays the foundation for my future group. In short, we… (1/4)

De-novo promoters emerge more readily from random DNA than from genomic DNA
Promoters are DNA sequences that help to initiate transcription. Point mutations can create de-novo promoters, which can consequently transcribe inactive genes or create novel transcripts. We know lit...
www.biorxiv.org
August 28, 2025 at 6:37 AM
Excited / nervous to share the “magnum opus” of my postdoc in Andreas Wagner’s lab!
"De-novo promoters emerge more readily from random DNA than from genomic DNA"
This project is the accumulation of 4 years of work, and lays the foundation for my future group. In short, we… (1/4)
"De-novo promoters emerge more readily from random DNA than from genomic DNA"
This project is the accumulation of 4 years of work, and lays the foundation for my future group. In short, we… (1/4)
Reposted by Jacob Hepkema

Activity of most genes is controlled by multiple enhancers, but is there activation coordinated? We leveraged Nanopore to identify a specific set of elements that are simultaneously accessible on the same DNA molecules and are coordinated in their activation. www.biorxiv.org/content/10.1...

August 18, 2025 at 12:23 PM
Activity of most genes is controlled by multiple enhancers, but is there activation coordinated? We leveraged Nanopore to identify a specific set of elements that are simultaneously accessible on the same DNA molecules and are coordinated in their activation. www.biorxiv.org/content/10.1...
Reposted by Jacob Hepkema
Interesting piece on automated enhancer design from the Seelig lab: www.cell.com/cell-systems...
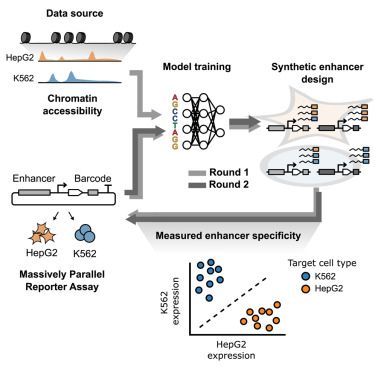
Iterative deep learning design of human enhancers exploits condensed sequence grammar to achieve cell-type specificity
Yin et al. demonstrate the use of iteratively retrained deep learning models to design
synthetic enhancers for progressively higher cell-type specificity. They show the
feasibility of model training f...
www.cell.com
July 15, 2025 at 6:19 AM
Interesting piece on automated enhancer design from the Seelig lab: www.cell.com/cell-systems...
Reposted by Jacob Hepkema

Our paper describing the Range Extender element which is required and sufficient for long-range enhancer activation at the Shh locus is now available at @nature.com. Congrats to @gracebower.bsky.social who led the study. Below is a brief summary of the main findings www.nature.com/articles/s41... 1/
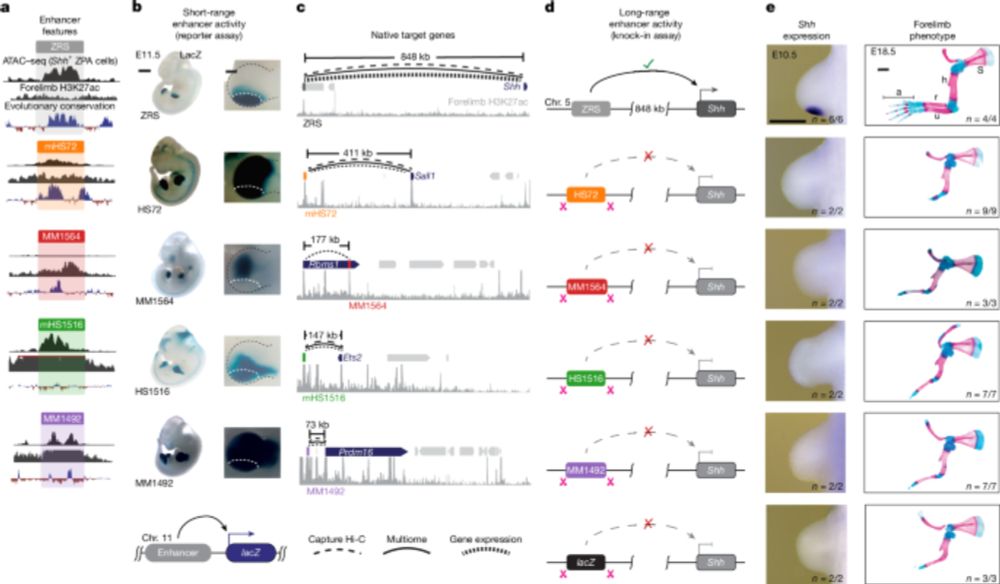
Range extender mediates long-distance enhancer activity - Nature
The REX element is associated with long-range enhancer–promoter interactions.
www.nature.com
July 2, 2025 at 4:17 PM
Our paper describing the Range Extender element which is required and sufficient for long-range enhancer activation at the Shh locus is now available at @nature.com. Congrats to @gracebower.bsky.social who led the study. Below is a brief summary of the main findings www.nature.com/articles/s41... 1/
Reposted by Jacob Hepkema

Textbooks: “Enhancers are just a bunch of TFBSs”
But how do they REALLY work?
New paper with many contributors here @berkeleylab.lbl.gov, @anshulkundaje.bsky.social, @anusri.bsky.social
A 🧵 (1/n)
Free access link: rdcu.be/erD22
But how do they REALLY work?
New paper with many contributors here @berkeleylab.lbl.gov, @anshulkundaje.bsky.social, @anusri.bsky.social
A 🧵 (1/n)
Free access link: rdcu.be/erD22
June 18, 2025 at 5:56 PM
Textbooks: “Enhancers are just a bunch of TFBSs”
But how do they REALLY work?
New paper with many contributors here @berkeleylab.lbl.gov, @anshulkundaje.bsky.social, @anusri.bsky.social
A 🧵 (1/n)
Free access link: rdcu.be/erD22
But how do they REALLY work?
New paper with many contributors here @berkeleylab.lbl.gov, @anshulkundaje.bsky.social, @anusri.bsky.social
A 🧵 (1/n)
Free access link: rdcu.be/erD22
Reposted by Jacob Hepkema

How to find Evolutionary Conserved Enhancers in 2025? 🐣-🐭
Check out our paper - fresh off the press!!!
We find widespread functional conservation of enhancers in absence of sequence homology
Including: a bioinformatic tool to map sequence-diverged enhancers!
rdcu.be/enVDN
github.com/tobiaszehnde...
Check out our paper - fresh off the press!!!
We find widespread functional conservation of enhancers in absence of sequence homology
Including: a bioinformatic tool to map sequence-diverged enhancers!
rdcu.be/enVDN
github.com/tobiaszehnde...
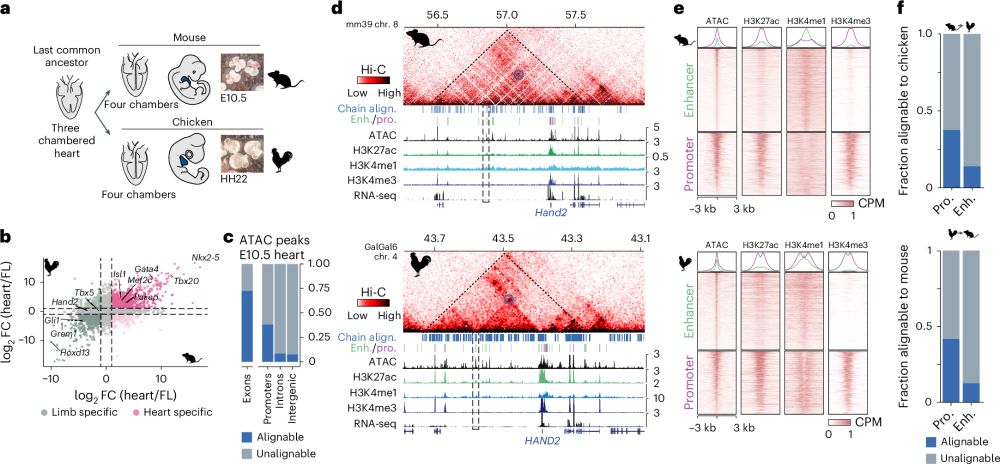
Conservation of regulatory elements with highly diverged sequences across large evolutionary distances
Nature Genetics - Combining functional genomic data from mouse and chicken with a synteny-based strategy identifies positionally conserved cis-regulatory elements in the absence of direct sequence...
rdcu.be
May 27, 2025 at 12:19 PM
How to find Evolutionary Conserved Enhancers in 2025? 🐣-🐭
Check out our paper - fresh off the press!!!
We find widespread functional conservation of enhancers in absence of sequence homology
Including: a bioinformatic tool to map sequence-diverged enhancers!
rdcu.be/enVDN
github.com/tobiaszehnde...
Check out our paper - fresh off the press!!!
We find widespread functional conservation of enhancers in absence of sequence homology
Including: a bioinformatic tool to map sequence-diverged enhancers!
rdcu.be/enVDN
github.com/tobiaszehnde...
Reposted by Jacob Hepkema

Our latest work now online in Cell:
Rewriting regulatory DNA to dissect and reprogram gene expression
Our new method (Variant-EFFECTS) uses high-throughput prime editing + flow sorting + sequencing to precisely measure effects of noncoding variants on gene expression
Thread 👇
Rewriting regulatory DNA to dissect and reprogram gene expression
Our new method (Variant-EFFECTS) uses high-throughput prime editing + flow sorting + sequencing to precisely measure effects of noncoding variants on gene expression
Thread 👇

April 17, 2025 at 6:26 PM
Our latest work now online in Cell:
Rewriting regulatory DNA to dissect and reprogram gene expression
Our new method (Variant-EFFECTS) uses high-throughput prime editing + flow sorting + sequencing to precisely measure effects of noncoding variants on gene expression
Thread 👇
Rewriting regulatory DNA to dissect and reprogram gene expression
Our new method (Variant-EFFECTS) uses high-throughput prime editing + flow sorting + sequencing to precisely measure effects of noncoding variants on gene expression
Thread 👇
Reposted by Jacob Hepkema

To the top of the "to-read" list. Looks like a heroic amount of work from the Hahn lab (large-scale ChEC-seq compendium!) www.nature.com/articles/s41...
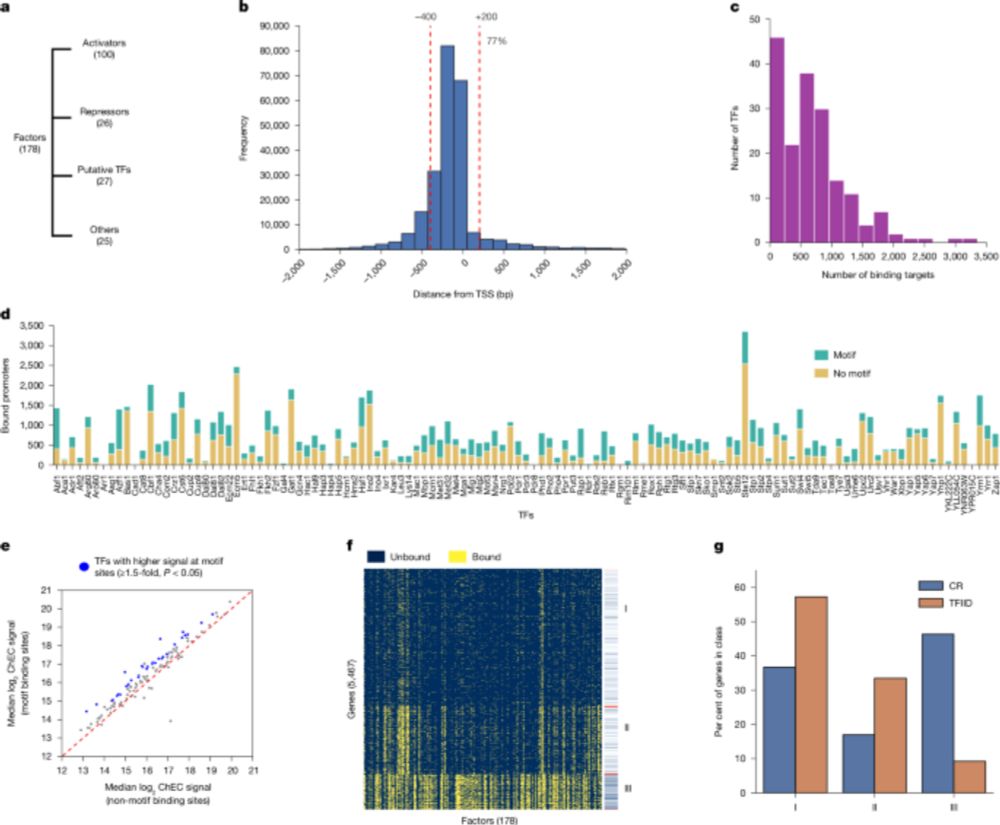
Low overlap of transcription factor DNA binding and regulatory targets - Nature
A near-complete survey of transcription factor activities in Saccharomyces cerevisiae reveals that most transcription factors have both activator and repressor activities and limited overlap between t...
www.nature.com
April 16, 2025 at 4:04 PM
To the top of the "to-read" list. Looks like a heroic amount of work from the Hahn lab (large-scale ChEC-seq compendium!) www.nature.com/articles/s41...
Reposted by Jacob Hepkema
A tour de force study from Taipale&Yin labs. It expands the vocabulary of the Regulatory Code by adding 1131 TF:TF composite motifs that are different from the individual TF motifs. The new composite motifs are enriched in cell-type specific elements and active in vivo
www.nature.com/articles/s41...
www.nature.com/articles/s41...
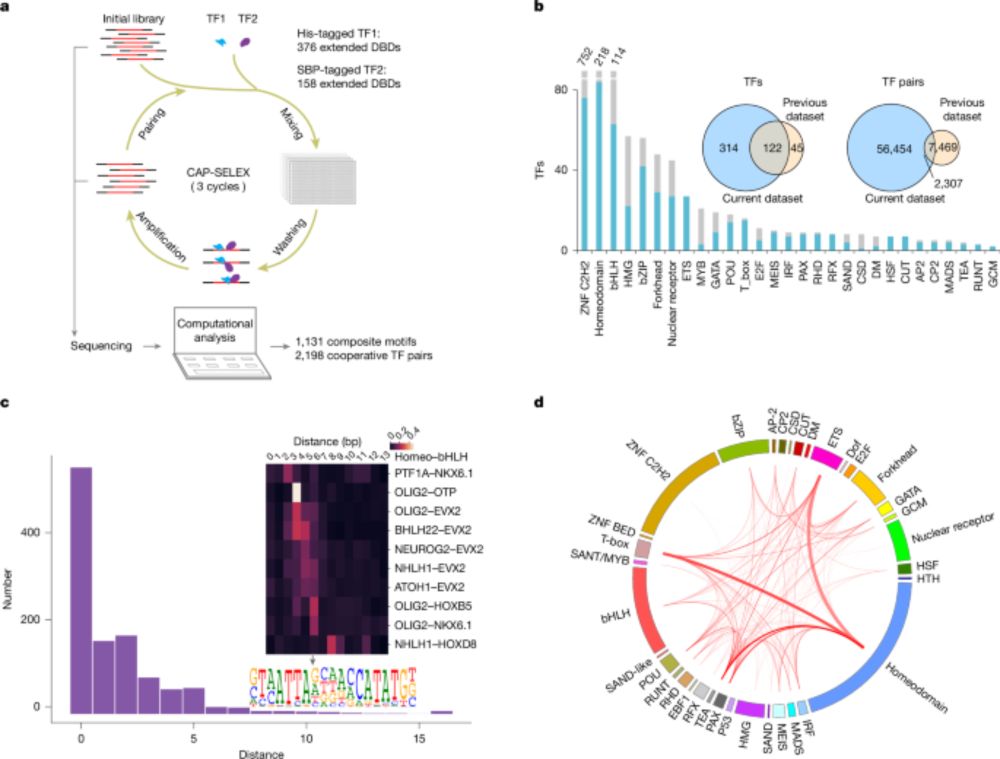
DNA-guided transcription factor interactions extend human gene regulatory code - Nature
A large-scale analysis of DNA-bound transcription factors (TFs) shows how the presence of DNA markedly affects the landscape of TF interactions, and identifies composite motifs that are recognized by ...
www.nature.com
April 9, 2025 at 4:51 PM
A tour de force study from Taipale&Yin labs. It expands the vocabulary of the Regulatory Code by adding 1131 TF:TF composite motifs that are different from the individual TF motifs. The new composite motifs are enriched in cell-type specific elements and active in vivo
www.nature.com/articles/s41...
www.nature.com/articles/s41...
Reposted by Jacob Hepkema

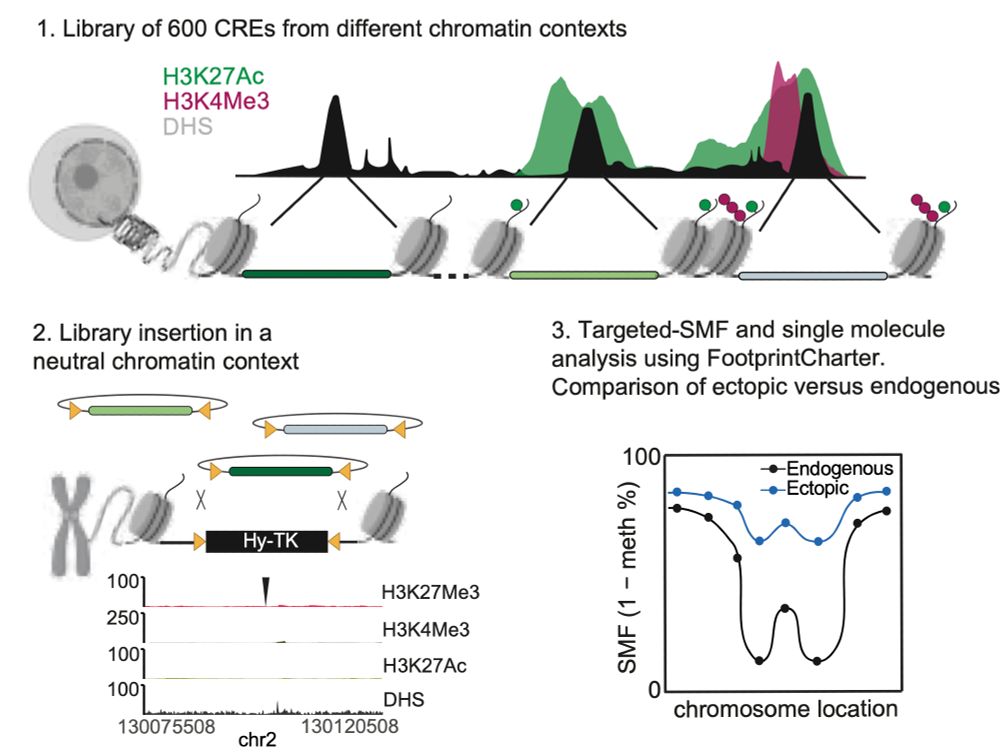
Happy to share the latest story from @arnaudkr.bsky.social's lab @embl.org! With @guidobarzaghi.bsky.social, we used Single Molecule Footprinting to quantify how often chromatin is accessible at enhancers after TF and chromatin environment changes! Check our preprint bit.ly/3XQMFxN + thread ⬇️ 1/11
April 8, 2025 at 1:52 PM
Happy to share the latest story from @arnaudkr.bsky.social's lab @embl.org! With @guidobarzaghi.bsky.social, we used Single Molecule Footprinting to quantify how often chromatin is accessible at enhancers after TF and chromatin environment changes! Check our preprint bit.ly/3XQMFxN + thread ⬇️ 1/11
Reposted by Jacob Hepkema

Our new preprint is out! Want to better visualize what your sequence-to-function profile learned? Here is PISA. It also comes in a new BPNet package, which can be used to train many genomics data sets, including MNase-seq data.

PISA: a versatile interpretation tool for visualizing cis-regulatory rules in genomic data https://www.biorxiv.org/content/10.1101/2025.04.07.647613v1
April 8, 2025 at 1:31 PM
Our new preprint is out! Want to better visualize what your sequence-to-function profile learned? Here is PISA. It also comes in a new BPNet package, which can be used to train many genomics data sets, including MNase-seq data.
Reposted by Jacob Hepkema
Very proud of two new preprints from the lab:
1) CREsted: to train sequence-to-function deep learning models on scATAC-seq atlases, and use them to decipher enhancer logic and design synthetic enhancers. This has been a wonderful lab-wide collaborative effort. www.biorxiv.org/content/10.1...
1) CREsted: to train sequence-to-function deep learning models on scATAC-seq atlases, and use them to decipher enhancer logic and design synthetic enhancers. This has been a wonderful lab-wide collaborative effort. www.biorxiv.org/content/10.1...

CREsted: modeling genomic and synthetic cell type-specific enhancers across tissues and species
Sequence-based deep learning models have become the state of the art for the analysis of the genomic regulatory code. Particularly for transcriptional enhancers, deep learning models excel at decipher...
www.biorxiv.org
April 4, 2025 at 9:04 AM
Very proud of two new preprints from the lab:
1) CREsted: to train sequence-to-function deep learning models on scATAC-seq atlases, and use them to decipher enhancer logic and design synthetic enhancers. This has been a wonderful lab-wide collaborative effort. www.biorxiv.org/content/10.1...
1) CREsted: to train sequence-to-function deep learning models on scATAC-seq atlases, and use them to decipher enhancer logic and design synthetic enhancers. This has been a wonderful lab-wide collaborative effort. www.biorxiv.org/content/10.1...
Reposted by Jacob Hepkema

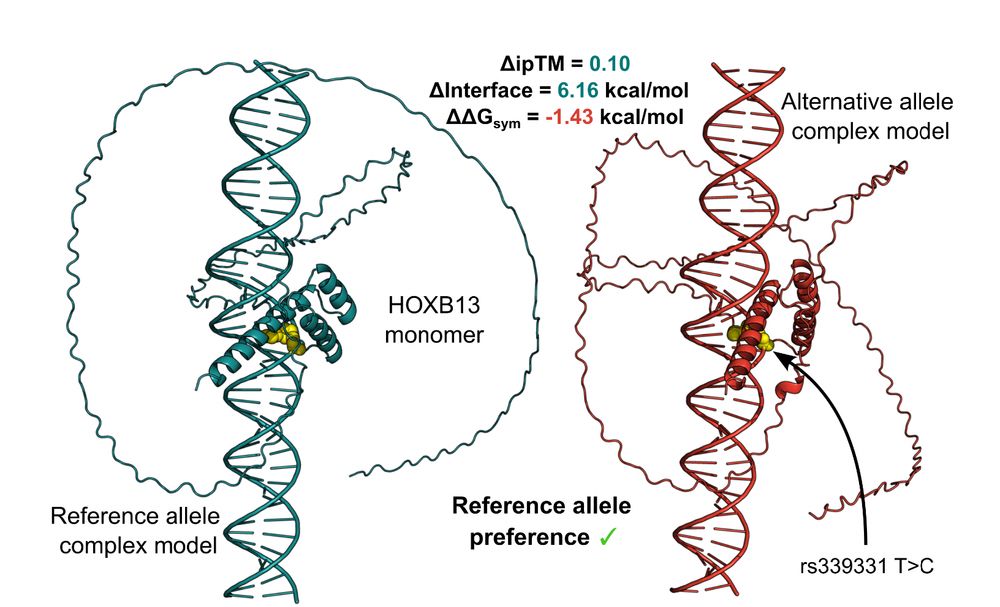
Unpicking non-coding genetic variation: Structure-guided modelling holds promise for evaluating how single nucleotide variants affect transcription factor binding. www.biorxiv.org/content/10.1.... @uoe-igc.bsky.social
March 21, 2025 at 10:22 AM
Unpicking non-coding genetic variation: Structure-guided modelling holds promise for evaluating how single nucleotide variants affect transcription factor binding. www.biorxiv.org/content/10.1.... @uoe-igc.bsky.social
Reposted by Jacob Hepkema

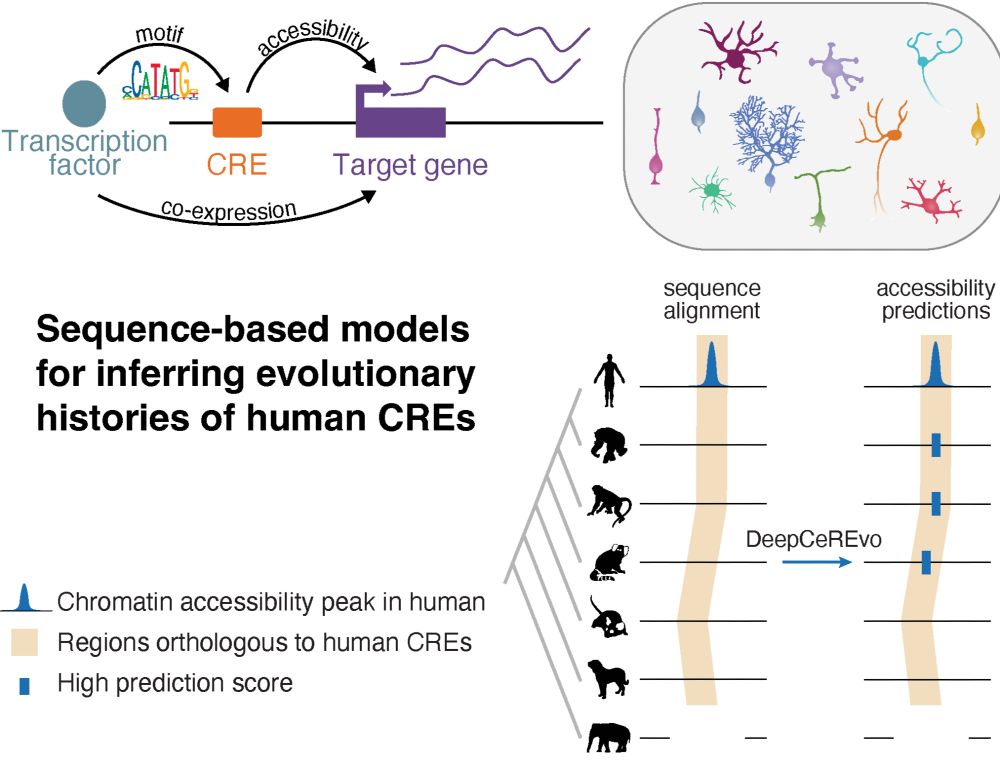
How does gene regulation shape brain evolution? Our new preprint dives into this question in the context of mammalian cerebellum development! rb.gy/dbcxjz
Led by @ioansarr.bsky.social, @marisepp.bsky.social and @tyamadat.bsky.social, in collaboration with @steinaerts.bsky.social
Led by @ioansarr.bsky.social, @marisepp.bsky.social and @tyamadat.bsky.social, in collaboration with @steinaerts.bsky.social
March 16, 2025 at 10:31 AM
How does gene regulation shape brain evolution? Our new preprint dives into this question in the context of mammalian cerebellum development! rb.gy/dbcxjz
Led by @ioansarr.bsky.social, @marisepp.bsky.social and @tyamadat.bsky.social, in collaboration with @steinaerts.bsky.social
Led by @ioansarr.bsky.social, @marisepp.bsky.social and @tyamadat.bsky.social, in collaboration with @steinaerts.bsky.social
Reposted by Jacob Hepkema

Just very happy to have our paper out today! A big thanks to all our co-authors, and to Nikolai and @steinaerts.bsky.social for the teamwork over the past years. If you are interested in using our models for cross-species enhancer studies, check out crested.readthedocs.io/en/stable/mo... 🙂
In a new study, Nikolai Hecker, Niklas Kempynck et al. in the team of @steinaerts.bsky.social explore 300 million years of brain evolution through the lens of enhancer codes.
www.science.org/doi/10.1126/...
www.science.org/doi/10.1126/...
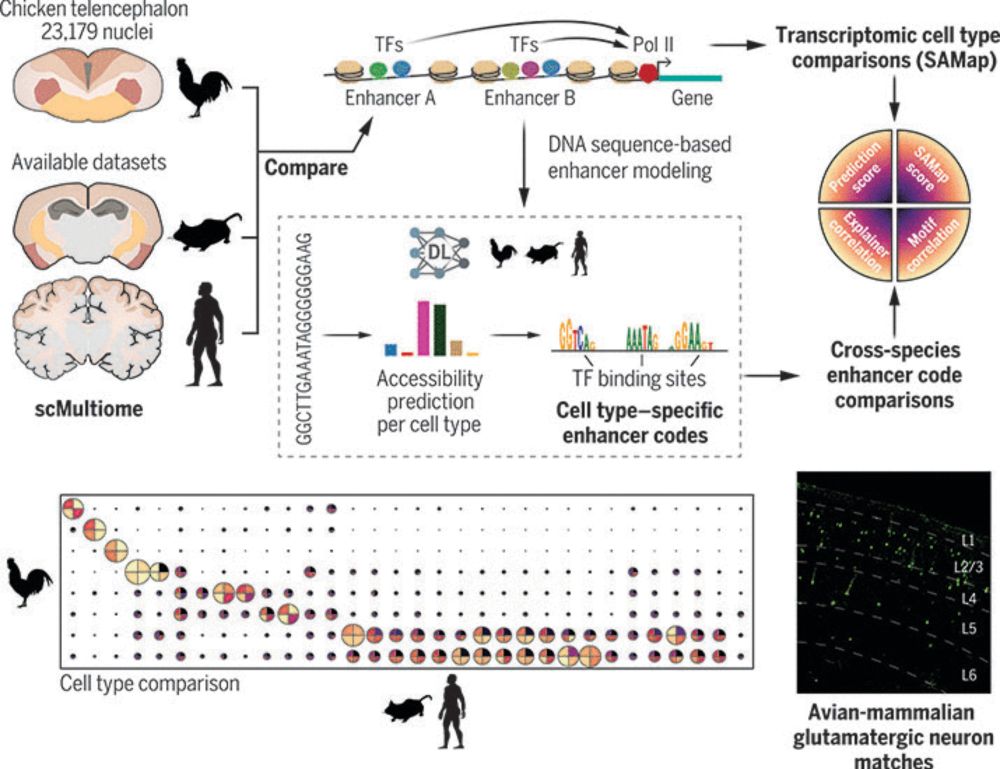
Enhancer-driven cell type comparison reveals similarities between the mammalian and bird pallium
Combinations of transcription factors govern the identity of cell types, which is reflected by genomic enhancer codes. We used deep learning to characterize these enhancer codes and devised three metr...
www.science.org
February 14, 2025 at 10:08 AM
Just very happy to have our paper out today! A big thanks to all our co-authors, and to Nikolai and @steinaerts.bsky.social for the teamwork over the past years. If you are interested in using our models for cross-species enhancer studies, check out crested.readthedocs.io/en/stable/mo... 🙂
Reposted by Jacob Hepkema

Recently, we've been playing around with using Ledidi to design "affinity catalogs" that exhibit a broad range of activities, rather than just the "most" of a desired activity.
Here are three example catalogs designed for GATA2 binding, chromatin accessibility, and transcription initiation.
Here are three example catalogs designed for GATA2 binding, chromatin accessibility, and transcription initiation.

February 12, 2025 at 12:17 PM
Recently, we've been playing around with using Ledidi to design "affinity catalogs" that exhibit a broad range of activities, rather than just the "most" of a desired activity.
Here are three example catalogs designed for GATA2 binding, chromatin accessibility, and transcription initiation.
Here are three example catalogs designed for GATA2 binding, chromatin accessibility, and transcription initiation.
Reposted by Jacob Hepkema

Our latest work: how can compartmentalization emerge in a eukaryotic genome lacking canonical heterochromatin?
By investigating bacterial genomes put in yeast, we show that the presence or absence of transcription is sufficient!
#chromatin #3Dgenome #generegulation
www.science.org/doi/10.1126/...
👇
By investigating bacterial genomes put in yeast, we show that the presence or absence of transcription is sufficient!
#chromatin #3Dgenome #generegulation
www.science.org/doi/10.1126/...
👇
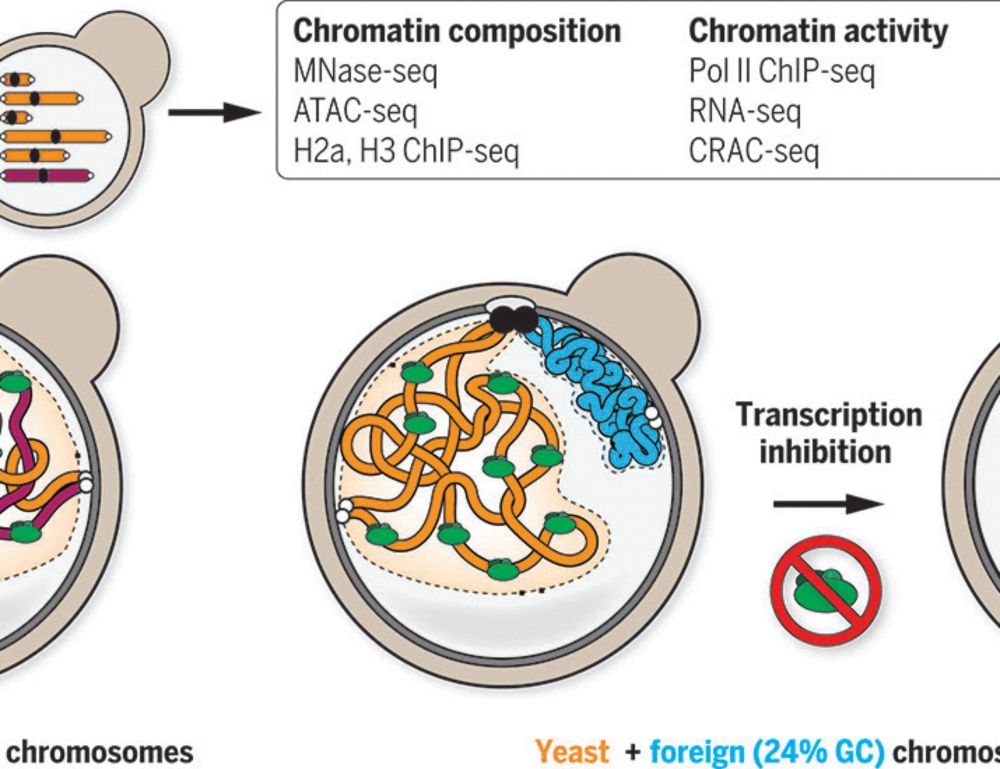
Sequence-dependent activity and compartmentalization of foreign DNA in a eukaryotic nucleus
In eukaryotes, DNA-associated protein complexes coevolve with genomic sequences to orchestrate chromatin folding. We investigate the relationship between DNA sequence and the spontaneous loading and a...
www.science.org
February 7, 2025 at 10:22 AM
Our latest work: how can compartmentalization emerge in a eukaryotic genome lacking canonical heterochromatin?
By investigating bacterial genomes put in yeast, we show that the presence or absence of transcription is sufficient!
#chromatin #3Dgenome #generegulation
www.science.org/doi/10.1126/...
👇
By investigating bacterial genomes put in yeast, we show that the presence or absence of transcription is sufficient!
#chromatin #3Dgenome #generegulation
www.science.org/doi/10.1126/...
👇
Reposted by Jacob Hepkema

Delighted to share our latest paper describing a method to read the levels of hundreds of metabolites or drugs in parallel using DNA sequencing. This method, which we call ‘smol-seq’ (Small MOLecule sequencing), harnesses the power of DNA sequencing for metabolite detection:
rdcu.be/d8xLv (1/6)
rdcu.be/d8xLv (1/6)
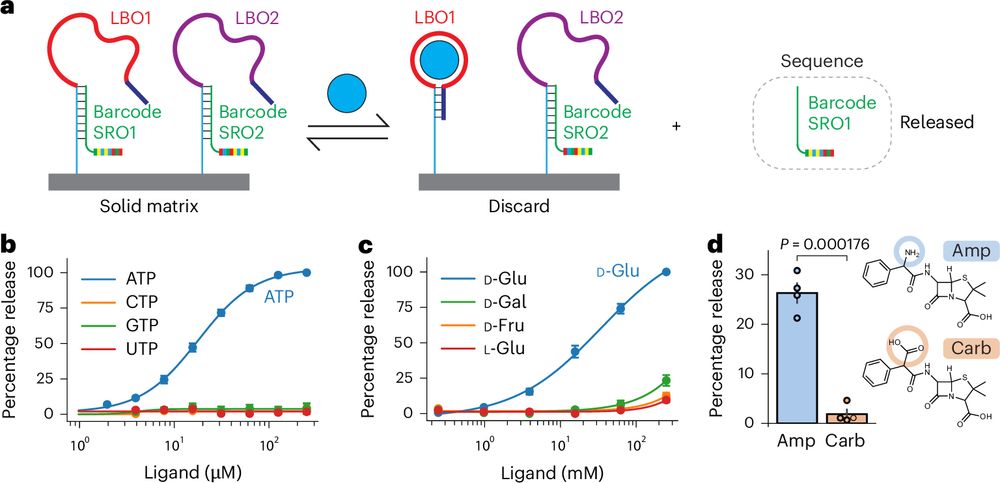
Quantifying metabolites using structure-switching aptamers coupled to DNA sequencing
Nature Biotechnology - Metabolites can be quantified using a combination of aptamers and DNA barcodes.
rdcu.be
February 4, 2025 at 1:54 PM
Delighted to share our latest paper describing a method to read the levels of hundreds of metabolites or drugs in parallel using DNA sequencing. This method, which we call ‘smol-seq’ (Small MOLecule sequencing), harnesses the power of DNA sequencing for metabolite detection:
rdcu.be/d8xLv (1/6)
rdcu.be/d8xLv (1/6)
Reposted by Jacob Hepkema

Now out in @science.org w/ @jshendure.bsky.social we present 'Genome-shuffle-seq': a method to shuffle mammalian genomes and characterize the impact of structural variants (SVs) with single-cell resolution in one experiment.
www.science.org/doi/10.1126/...
www.science.org/doi/10.1126/...
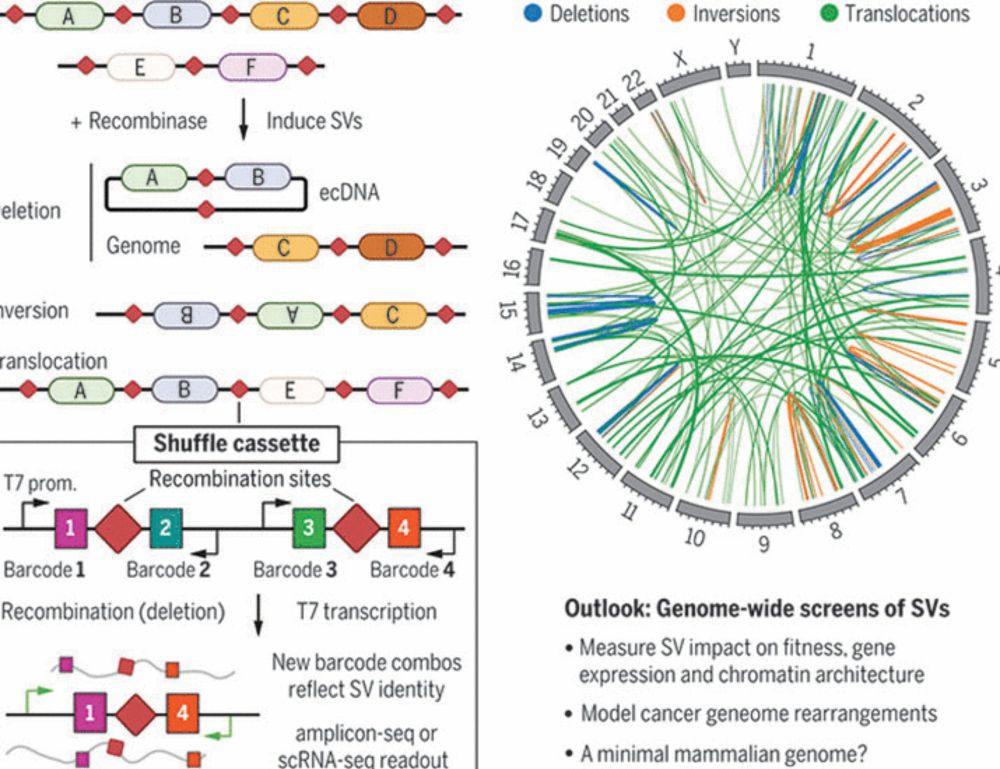
Multiplex generation and single-cell analysis of structural variants in mammalian genomes
Studying the functional consequences of structural variants (SVs) in mammalian genomes is challenging because (i) SVs arise much less commonly than single-nucleotide variants or small indels and (ii) ...
www.science.org
January 31, 2025 at 7:41 PM
Now out in @science.org w/ @jshendure.bsky.social we present 'Genome-shuffle-seq': a method to shuffle mammalian genomes and characterize the impact of structural variants (SVs) with single-cell resolution in one experiment.
www.science.org/doi/10.1126/...
www.science.org/doi/10.1126/...
Reposted by Jacob Hepkema
We're delighted to share our work on scrambling the human genome using prime editing, repetitive elements, and recombinases in @science.org , led by @jonaskoeppel.bsky.social , @f-raphael.bsky.social , with @proftomellis.bsky.social and George Church.
www.science.org/doi/10.1126/...
www.science.org/doi/10.1126/...
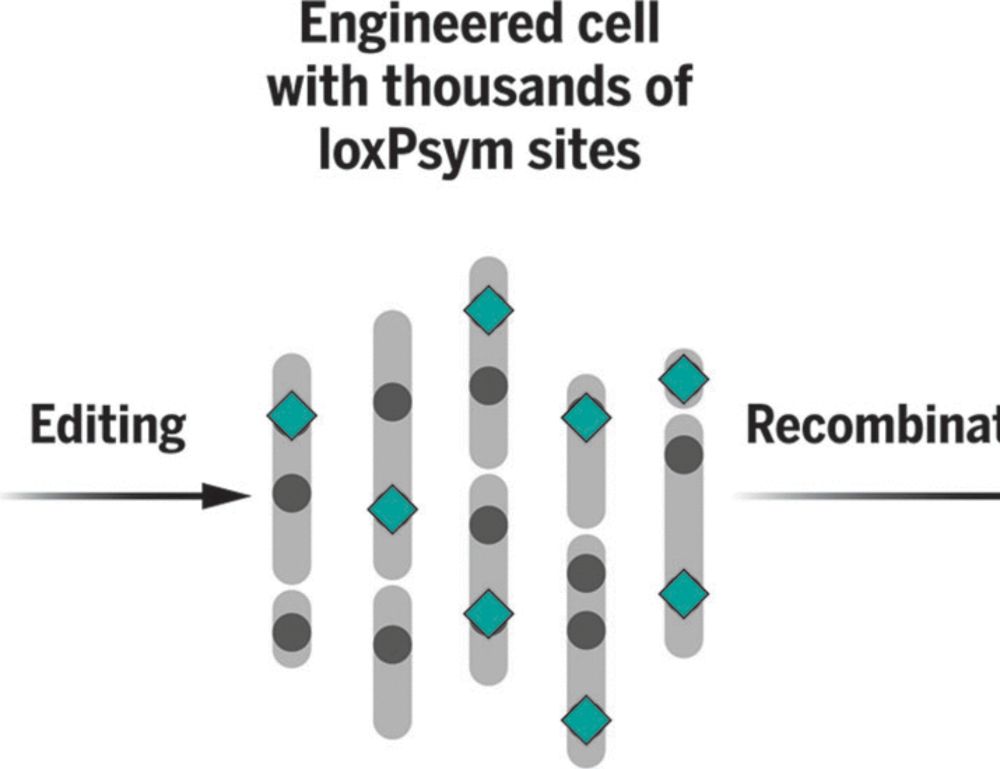
Randomizing the human genome by engineering recombination between repeat elements
We lack tools to edit DNA sequences at scales necessary to study 99% of the human genome that is noncoding. To address this gap, we applied CRISPR prime editing to insert recombination handles into re...
www.science.org
January 31, 2025 at 1:49 PM
We're delighted to share our work on scrambling the human genome using prime editing, repetitive elements, and recombinases in @science.org , led by @jonaskoeppel.bsky.social , @f-raphael.bsky.social , with @proftomellis.bsky.social and George Church.
www.science.org/doi/10.1126/...
www.science.org/doi/10.1126/...
Reposted by Jacob Hepkema

0/ Essential reading for anyone training or using sequence-function models trained on genomic sequences! 🚨 In our new preprint, we explore the ways homology within genomes can cause leakage when training sequence-based models and ways to prevent it
January 27, 2025 at 11:04 PM
0/ Essential reading for anyone training or using sequence-function models trained on genomic sequences! 🚨 In our new preprint, we explore the ways homology within genomes can cause leakage when training sequence-based models and ways to prevent it
Looks like a very useful method for deeper chromatin accessibility analysis!

Super excited to share our new study from the @jbuenrostro.bsky.social Lab in @nature.com! We developed a computational method for tracking transcription factor and nucleosome binding using single-cell ATAC-seq and deep learning.
Paper: www.nature.com/articles/s41...
Paper: www.nature.com/articles/s41...
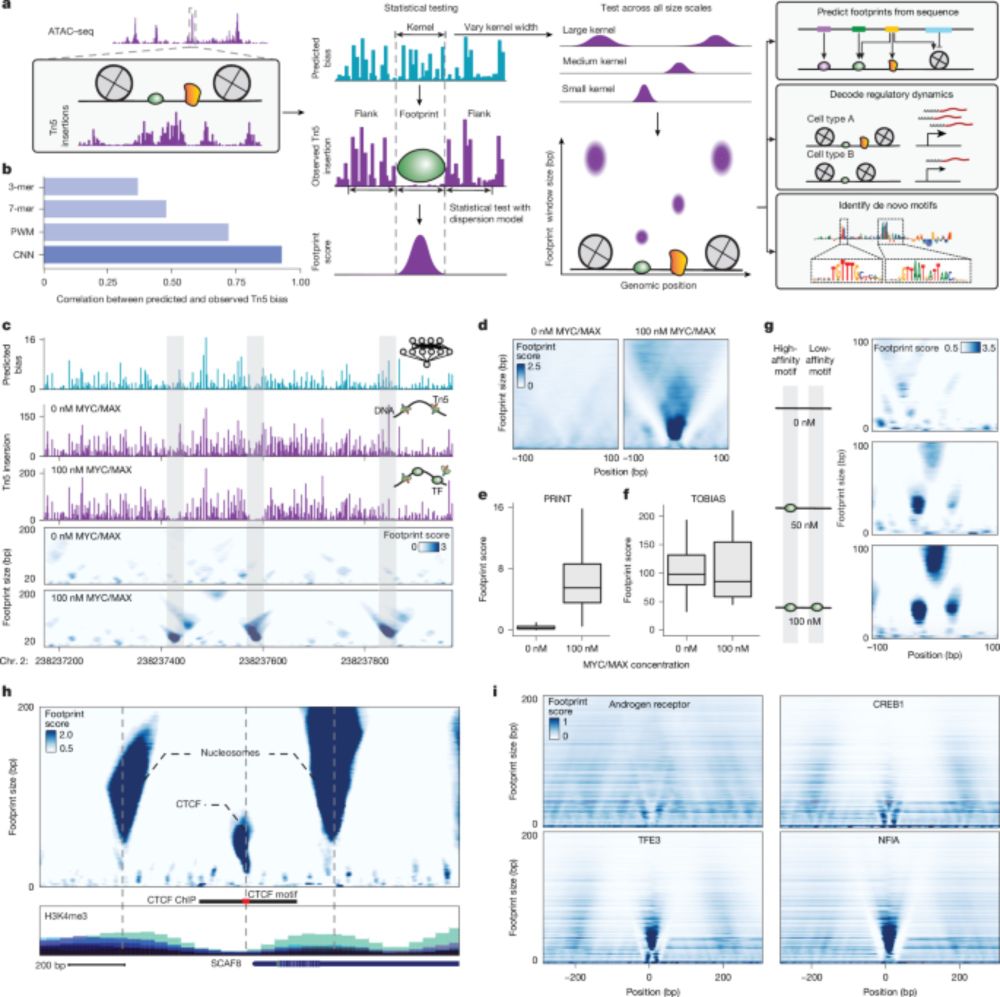
Multiscale footprints reveal the organization of cis-regulatory elements - Nature
We developed PRINT, a computational method that identifies footprints of DNA–protein interactions from bulk and single-cell chromatin accessibility data across multiple scales of protein size.
www.nature.com
January 23, 2025 at 7:34 AM
Looks like a very useful method for deeper chromatin accessibility analysis!