


Paul Medvedev
@pashadag.bsky.social
Algorithmic Bioinformatics Researcher and Teacher. Posts about research results and educational/mentorship topics (for details, see http://bit.ly/380vX22).
Reposted by Paul Medvedev

Interested in a post-doc in Israel? The deadline for the Azrieli International Postdoctoral Fellowship is November 19. The fellowship offers generous funding for postdocs to conduct research in any academic discipline at eligible Israeli institutions: azrielifoundation.org/fellows/inte...
International Postdoctoral Fellowship - The Azrieli Foundation
The Azrieli Fellows Program is an elite group of academics who cultivate a network of leading professionals in Israel and around the world.
azrielifoundation.org
November 10, 2025 at 8:59 AM
Interested in a post-doc in Israel? The deadline for the Azrieli International Postdoctoral Fellowship is November 19. The fellowship offers generous funding for postdocs to conduct research in any academic discipline at eligible Israeli institutions: azrielifoundation.org/fellows/inte...
Reposted by Paul Medvedev

Haonan Wu gives a talk on "A k-mer-based estimator of the substitution rate between repetitive sequences"
www.biorxiv.org/content/10.1...
This work tackles the issue of Mash which ignores repeats in the genome, providing better distance estimation #GI2025
www.biorxiv.org/content/10.1...
This work tackles the issue of Mash which ignores repeats in the genome, providing better distance estimation #GI2025


November 6, 2025 at 4:38 PM
Haonan Wu gives a talk on "A k-mer-based estimator of the substitution rate between repetitive sequences"
www.biorxiv.org/content/10.1...
This work tackles the issue of Mash which ignores repeats in the genome, providing better distance estimation #GI2025
www.biorxiv.org/content/10.1...
This work tackles the issue of Mash which ignores repeats in the genome, providing better distance estimation #GI2025
Reposted by Paul Medvedev
After years of research and continuous refinement, we’re thrilled to share that our paper on the MetaGraph framework — enabling Petabase-scale search across sequencing data — has been published today in Nature (www.nature.com/articles/s41...)

Efficient and accurate search in petabase-scale sequence repositories - Nature
MetaGraph enables scalable indexing of large sets of DNA, RNA or protein sequences using annotated de Bruijn graphs.
www.nature.com
October 8, 2025 at 8:56 PM
After years of research and continuous refinement, we’re thrilled to share that our paper on the MetaGraph framework — enabling Petabase-scale search across sequencing data — has been published today in Nature (www.nature.com/articles/s41...)
Reposted by Paul Medvedev

And it's posted! If you're interested and eligible, please consider applying through the UMD portal: umd.wd1.myworkdayjobs.com/en-US/UMCP/j....
If you're a PI working in algorithmic genomics (& you can recommend my lab to your top graduating students ;P), please let them know!
If you're a PI working in algorithmic genomics (& you can recommend my lab to your top graduating students ;P), please let them know!
October 8, 2025 at 4:53 PM
And it's posted! If you're interested and eligible, please consider applying through the UMD portal: umd.wd1.myworkdayjobs.com/en-US/UMCP/j....
If you're a PI working in algorithmic genomics (& you can recommend my lab to your top graduating students ;P), please let them know!
If you're a PI working in algorithmic genomics (& you can recommend my lab to your top graduating students ;P), please let them know!
Reposted by Paul Medvedev

I've added 7 videos to my Burrows-Wheeler indexing playlist (www.youtube.com/playlist?lis...), rounding out the r-index series and adding a 5-part series on the move structure. Now 27 videos in that playlist. I aim to add videos on prefix-free parsing, PBWT, Wheeler languages/automata in the future.

Burrows-Wheeler Indexing - YouTube
Videos on : (a) the Burrows-Wheeler Transform (BWT), (b) the FM Index, which uses the BWT to construct a full-text index, (c) Wheeler graphs, (d) r-index, an...
www.youtube.com
October 7, 2025 at 2:17 PM
I've added 7 videos to my Burrows-Wheeler indexing playlist (www.youtube.com/playlist?lis...), rounding out the r-index series and adding a 5-part series on the move structure. Now 27 videos in that playlist. I aim to add videos on prefix-free parsing, PBWT, Wheeler languages/automata in the future.
Reposted by Paul Medvedev

Our preprint on our new metagenomic HiFi assembler Alice is out 🥳 Based on a *new sketching method* (🧵1/6)
👉 Preprint www.biorxiv.org/content/10.1...
👉 Github github.com/rolandfaure/...
👉 Preprint www.biorxiv.org/content/10.1...
👉 Github github.com/rolandfaure/...

Alice: fast and haplotype-aware assembly of high-fidelity reads based on MSR sketching
We introduce Mapping-friendly Sequence Reduction (MSR) sketches, a sketching method for high-fidelity (HiFi) long reads, and Alice, an assembler that operates directly on these sketches. MSR produces ...
www.biorxiv.org
October 3, 2025 at 2:51 PM
Our preprint on our new metagenomic HiFi assembler Alice is out 🥳 Based on a *new sketching method* (🧵1/6)
👉 Preprint www.biorxiv.org/content/10.1...
👉 Github github.com/rolandfaure/...
👉 Preprint www.biorxiv.org/content/10.1...
👉 Github github.com/rolandfaure/...
Reposted by Paul Medvedev

Alice: fast and haplotype-aware assembly of high-fidelity reads based on MSR sketching https://www.biorxiv.org/content/10.1101/2025.09.29.679204v1
October 1, 2025 at 1:47 AM
Alice: fast and haplotype-aware assembly of high-fidelity reads based on MSR sketching https://www.biorxiv.org/content/10.1101/2025.09.29.679204v1
Reposted by Paul Medvedev

#RECOMB2026 will be in Thessaloniki, Greece on May 26-29, 2026. Satellites on May 24-25. Save the date!
Το συνέδριο #RECOMB2026 θα πραγματοποιηθεί στη Θεσσαλονίκη, στις 26-29 Μαΐου 2026. Οι δορυφορικές εκδηλώσεις θα διεξαχθούν στις 24-25 Μαΐου 2026. Σημειώστε την ημερομηνία!
Το συνέδριο #RECOMB2026 θα πραγματοποιηθεί στη Θεσσαλονίκη, στις 26-29 Μαΐου 2026. Οι δορυφορικές εκδηλώσεις θα διεξαχθούν στις 24-25 Μαΐου 2026. Σημειώστε την ημερομηνία!

September 26, 2025 at 3:03 PM
#RECOMB2026 will be in Thessaloniki, Greece on May 26-29, 2026. Satellites on May 24-25. Save the date!
Το συνέδριο #RECOMB2026 θα πραγματοποιηθεί στη Θεσσαλονίκη, στις 26-29 Μαΐου 2026. Οι δορυφορικές εκδηλώσεις θα διεξαχθούν στις 24-25 Μαΐου 2026. Σημειώστε την ημερομηνία!
Το συνέδριο #RECOMB2026 θα πραγματοποιηθεί στη Θεσσαλονίκη, στις 26-29 Μαΐου 2026. Οι δορυφορικές εκδηλώσεις θα διεξαχθούν στις 24-25 Μαΐου 2026. Σημειώστε την ημερομηνία!
If you're wondering why we're hosting the pre-print via dropbox, its because arXiv (and bioRxiv) did not accept it (because it is a review). Its a bit disconcerting, because a review is precisely the type of paper that would benefit a lot from pre-publication dissemination and feedback.
Thank you folks for your feedback on our survey about Hash functions in genomic sequence analysis. We've updated the paper and you can see the new version here: tinyurl.com/4kk9ccmt.
September 25, 2025 at 1:25 PM
If you're wondering why we're hosting the pre-print via dropbox, its because arXiv (and bioRxiv) did not accept it (because it is a review). Its a bit disconcerting, because a review is precisely the type of paper that would benefit a lot from pre-publication dissemination and feedback.
Thank you folks for your feedback on our survey about Hash functions in genomic sequence analysis. We've updated the paper and you can see the new version here: tinyurl.com/4kk9ccmt.
September 25, 2025 at 1:21 PM
Thank you folks for your feedback on our survey about Hash functions in genomic sequence analysis. We've updated the paper and you can see the new version here: tinyurl.com/4kk9ccmt.
Reposted by Paul Medvedev
Excited to share our EvANI benchmarking workflow, published in Briefings in Bioinformatics doi.org/10.1093/bib/...
Computing average nucleotide identity (ANI) is neither conceptually nor computationally trivial. Its definition has evolved over years, with different meanings and assumptions (1/5)
Computing average nucleotide identity (ANI) is neither conceptually nor computationally trivial. Its definition has evolved over years, with different meanings and assumptions (1/5)

September 21, 2025 at 3:26 PM
Excited to share our EvANI benchmarking workflow, published in Briefings in Bioinformatics doi.org/10.1093/bib/...
Computing average nucleotide identity (ANI) is neither conceptually nor computationally trivial. Its definition has evolved over years, with different meanings and assumptions (1/5)
Computing average nucleotide identity (ANI) is neither conceptually nor computationally trivial. Its definition has evolved over years, with different meanings and assumptions (1/5)
Reposted by Paul Medvedev

Preprint out for myloasm, our new nanopore / HiFi metagenome assembler!
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
High-resolution metagenome assembly for modern long reads with myloasm https://www.biorxiv.org/content/10.1101/2025.09.05.674543v1
September 7, 2025 at 11:35 PM
Preprint out for myloasm, our new nanopore / HiFi metagenome assembler!
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
Reposted by Paul Medvedev

🌎👩🔬 For 15+ years biology has accumulated petabytes (million gigabytes) of🧬DNA sequencing data🧬 from the far reaches of our planet.🦠🍄🌵
Logan now democratizes efficient access to the world’s most comprehensive genetics dataset. Free and open.
doi.org/10.1101/2024...
Logan now democratizes efficient access to the world’s most comprehensive genetics dataset. Free and open.
doi.org/10.1101/2024...

September 3, 2025 at 8:39 AM
🌎👩🔬 For 15+ years biology has accumulated petabytes (million gigabytes) of🧬DNA sequencing data🧬 from the far reaches of our planet.🦠🍄🌵
Logan now democratizes efficient access to the world’s most comprehensive genetics dataset. Free and open.
doi.org/10.1101/2024...
Logan now democratizes efficient access to the world’s most comprehensive genetics dataset. Free and open.
doi.org/10.1101/2024...
Reposted by Paul Medvedev

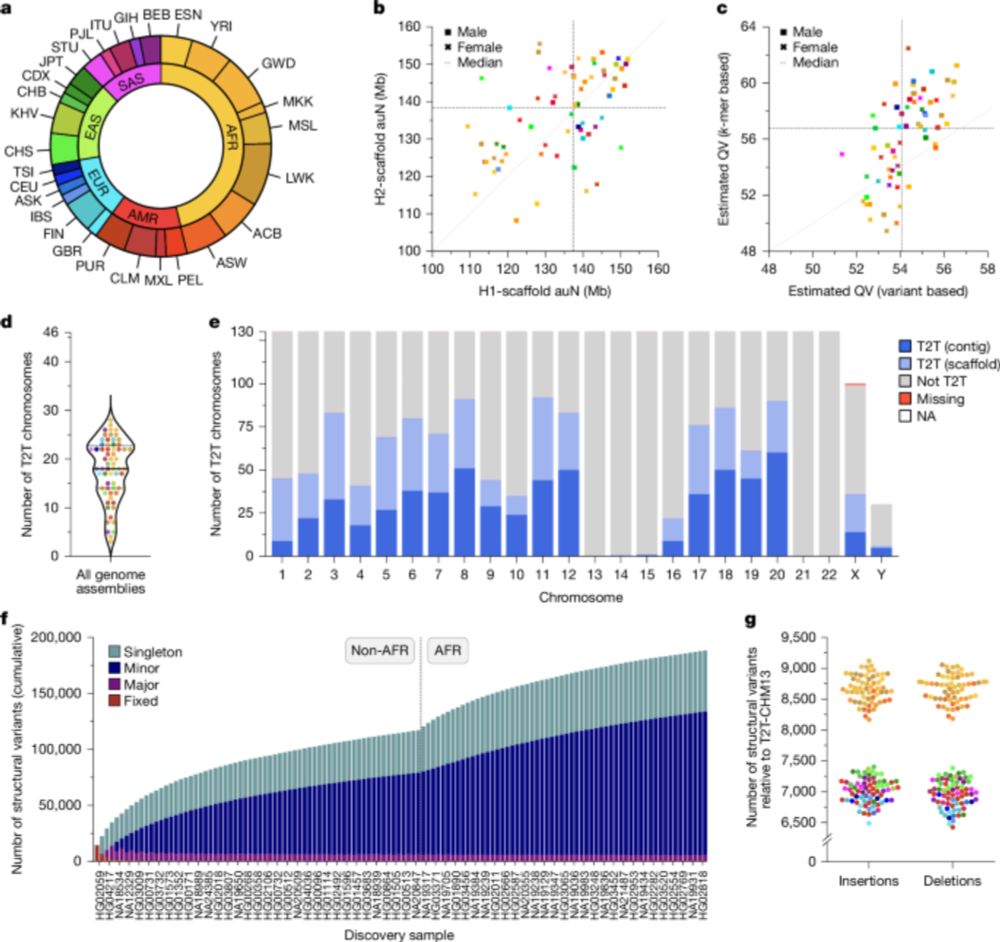
Two papers in today's issue of @nature.com : 1) we assemble 65 genomes to near completion, including centromeres and the MHC. tinyurl.com/3huhax6w. 2) we sequence 1,019 genomes from the 1kGP with long reads, revealing SVs down to low allele frequencies tinyurl.com/wbx3we9x.
Complex genetic variation in nearly complete human genomes - Nature
Using sequencing and haplotype-resolved assembly of 65 diverse human genomes, complex regions including the major histocompatibility complex and centromeres are analysed.
tinyurl.com
July 23, 2025 at 3:12 PM
Two papers in today's issue of @nature.com : 1) we assemble 65 genomes to near completion, including centromeres and the MHC. tinyurl.com/3huhax6w. 2) we sequence 1,019 genomes from the 1kGP with long reads, revealing SVs down to low allele frequencies tinyurl.com/wbx3we9x.
Reposted by Paul Medvedev

Interested in a tool that aligns millions of proteins in minutes with quality similar to or better than the state-of-the-art utilities? Please take a look at our FAMSA2 paper: www.biorxiv.org/content/10.1...
and GH repo: github.com/refresh-bio/...
and GH repo: github.com/refresh-bio/...

FAMSA2 enables accurate multiple sequence alignment at protein-universe scale
We introduce FAMSA2, an algorithm that produces high-accuracy multiple protein sequence alignments with unprecedented speed. Across structural, phylogenetic, and functional benchmarks, FAMSA2 matches ...
www.biorxiv.org
July 19, 2025 at 9:28 PM
Interested in a tool that aligns millions of proteins in minutes with quality similar to or better than the state-of-the-art utilities? Please take a look at our FAMSA2 paper: www.biorxiv.org/content/10.1...
and GH repo: github.com/refresh-bio/...
and GH repo: github.com/refresh-bio/...
Reposted by Paul Medvedev
Sassy: Searching Short DNA Strings in the 2020s https://www.biorxiv.org/content/10.1101/2025.07.22.666207v1
July 26, 2025 at 6:46 PM
Sassy: Searching Short DNA Strings in the 2020s https://www.biorxiv.org/content/10.1101/2025.07.22.666207v1
Reposted by Paul Medvedev

Congratulations to Rayan Chiki, (Institut Pasteur) head of the “Sequence Bioinformatics” unit, for securing the ERC Proof of Concept 2025 for his project ENZYMINER! 👏
@rayan.chiki.bsky.social
#Bioinformatics
@rayan.chiki.bsky.social
#Bioinformatics


July 24, 2025 at 3:10 PM
Congratulations to Rayan Chiki, (Institut Pasteur) head of the “Sequence Bioinformatics” unit, for securing the ERC Proof of Concept 2025 for his project ENZYMINER! 👏
@rayan.chiki.bsky.social
#Bioinformatics
@rayan.chiki.bsky.social
#Bioinformatics
Reposted by Paul Medvedev

After Tim Hunt won the Nobel, he said, "We do science because we like discovering things about the world...and then boasting about what we found".
Any one individual can argue about their own motivation, but it would naive to dispute that's an accurate description of many people.
Any one individual can argue about their own motivation, but it would naive to dispute that's an accurate description of many people.
July 11, 2025 at 1:44 PM
After Tim Hunt won the Nobel, he said, "We do science because we like discovering things about the world...and then boasting about what we found".
Any one individual can argue about their own motivation, but it would naive to dispute that's an accurate description of many people.
Any one individual can argue about their own motivation, but it would naive to dispute that's an accurate description of many people.
Reposted by Paul Medvedev

Paper Alert!
Our preprint on the K2R index, being able to efficiently associate kmers to the reads containing them is finally out there!
A thread!
academic.oup.com/bioinformati...
Our preprint on the K2R index, being able to efficiently associate kmers to the reads containing them is finally out there!
A thread!
academic.oup.com/bioinformati...
K2R: Tinted de Bruijn graphs implementation for efficient read extraction from sequencing datasets
AbstractSummary. Biological sequence analysis often relies on reference genomes, but producing accurate assemblies remains a challenge. As a result, de nov
academic.oup.com
July 5, 2025 at 9:31 AM
Paper Alert!
Our preprint on the K2R index, being able to efficiently associate kmers to the reads containing them is finally out there!
A thread!
academic.oup.com/bioinformati...
Our preprint on the K2R index, being able to efficiently associate kmers to the reads containing them is finally out there!
A thread!
academic.oup.com/bioinformati...
Reposted by Paul Medvedev
Paper alert!
We present Oreo a tools that reorder long reads datasets in a way to compress them efficiently with ANY universal compressor like gz, zstd, xz ...
TLDR: You can get state of the art compression WITHOUT a dedicated compressor/decompressor!
academic.oup.com/bioinformati...
A thread!
We present Oreo a tools that reorder long reads datasets in a way to compress them efficiently with ANY universal compressor like gz, zstd, xz ...
TLDR: You can get state of the art compression WITHOUT a dedicated compressor/decompressor!
academic.oup.com/bioinformati...
A thread!
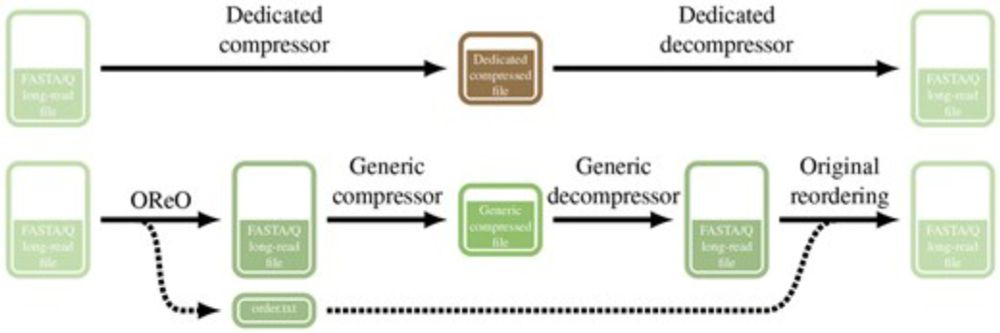
OReO: optimizing read order for practical compression
AbstractMotivation. Recent advances in high-throughput and third-generation sequencing technologies have created significant challenges in storing and mana
academic.oup.com
July 3, 2025 at 10:53 AM
Paper alert!
We present Oreo a tools that reorder long reads datasets in a way to compress them efficiently with ANY universal compressor like gz, zstd, xz ...
TLDR: You can get state of the art compression WITHOUT a dedicated compressor/decompressor!
academic.oup.com/bioinformati...
A thread!
We present Oreo a tools that reorder long reads datasets in a way to compress them efficiently with ANY universal compressor like gz, zstd, xz ...
TLDR: You can get state of the art compression WITHOUT a dedicated compressor/decompressor!
academic.oup.com/bioinformati...
A thread!
Reposted by Paul Medvedev

I worked with Thomas during a three months research visit during his PhD, and it resulted in a paper in NAR. I highly recommend him. doi.org/10.1093/nar/...
July 2, 2025 at 11:48 AM
I worked with Thomas during a three months research visit during his PhD, and it resulted in a paper in NAR. I highly recommend him. doi.org/10.1093/nar/...
Reposted by Paul Medvedev
Preprint alert!
We present K2Rmini, an ultra-fast, grep-like tool that extracts sequences of interest from FASTA/FASTQ files based on their k-mer content.
www.biorxiv.org/content/10.1...
A thread
We present K2Rmini, an ultra-fast, grep-like tool that extracts sequences of interest from FASTA/FASTQ files based on their k-mer content.
www.biorxiv.org/content/10.1...
A thread

Accelerating k-mer-based sequence filtering
The exponential growth of global sequencing data repositories presents both analytical challenges and opportunities. While k - mer-based indexing has improved scalability over traditional alignment fo...
www.biorxiv.org
July 2, 2025 at 1:00 PM
Preprint alert!
We present K2Rmini, an ultra-fast, grep-like tool that extracts sequences of interest from FASTA/FASTQ files based on their k-mer content.
www.biorxiv.org/content/10.1...
A thread
We present K2Rmini, an ultra-fast, grep-like tool that extracts sequences of interest from FASTA/FASTQ files based on their k-mer content.
www.biorxiv.org/content/10.1...
A thread
Reposted by Paul Medvedev
🖥️🧬 WABI '25 will not only have excellent keynotes, but an exciting program of papers. The titles and abstracts of all accepted WABI '25 papers are now available on the conference website (wabiconf.github.io/2025/talks/). I'm looking forward to seeing these talks!
June 25, 2025 at 6:40 PM
🖥️🧬 WABI '25 will not only have excellent keynotes, but an exciting program of papers. The titles and abstracts of all accepted WABI '25 papers are now available on the conference website (wabiconf.github.io/2025/talks/). I'm looking forward to seeing these talks!
🧵1/n
Estimating mutation rates using k-mers is fast—but what happens when repeats dominate the genome?
In a new preprint, Haonan Wu, Antonio Blanca, and myself propose a *repeat-aware* estimator that's accurate even in centromeres.
Estimating mutation rates using k-mers is fast—but what happens when repeats dominate the genome?
In a new preprint, Haonan Wu, Antonio Blanca, and myself propose a *repeat-aware* estimator that's accurate even in centromeres.
A k-mer-based estimator of the substitution rate between repetitive sequences https://www.biorxiv.org/content/10.1101/2025.06.19.660607v1
June 25, 2025 at 1:19 PM
🧵1/n
Estimating mutation rates using k-mers is fast—but what happens when repeats dominate the genome?
In a new preprint, Haonan Wu, Antonio Blanca, and myself propose a *repeat-aware* estimator that's accurate even in centromeres.
Estimating mutation rates using k-mers is fast—but what happens when repeats dominate the genome?
In a new preprint, Haonan Wu, Antonio Blanca, and myself propose a *repeat-aware* estimator that's accurate even in centromeres.