



Roland Faure
@rfaure.bsky.social
Sequence bioinfomatician, algorithms, methods.
Postdoc in Institut Pasteur in Rayan Chikhi's lab
Postdoc in Institut Pasteur in Rayan Chikhi's lab
Reposted by Roland Faure

"..based on a common wavefront design that can be adapted to support a variety of dynamic programming algorithms: local, global, and semi-global alignment of genomic and protein sequences with a variety of commonly used scoring schemes" from
@martinsteinegger.bsky.social andco
@martinsteinegger.bsky.social andco
Accelign: a GPU-based Library for Accelerating Pairwise Sequence Alignment https://www.biorxiv.org/content/10.64898/2025.12.17.694868v1
December 20, 2025 at 11:05 AM
"..based on a common wavefront design that can be adapted to support a variety of dynamic programming algorithms: local, global, and semi-global alignment of genomic and protein sequences with a variety of commonly used scoring schemes" from
@martinsteinegger.bsky.social andco
@martinsteinegger.bsky.social andco
Reposted by Roland Faure

Inverted colored de Bruijn Graph for practical kmer sets storage https://www.biorxiv.org/content/10.64898/2025.12.08.692073v1
December 11, 2025 at 12:46 AM
Inverted colored de Bruijn Graph for practical kmer sets storage https://www.biorxiv.org/content/10.64898/2025.12.08.692073v1
Reposted by Roland Faure

The 12th edition of the 2-days workshop “Data Structures in Bioinformatics” (DSB) will take place in Venice (Italy) on February 18-19th, 2026: dsb-meeting.github.io/DSB2026/
DSB 2026 Venice - February 18-19
Workshop Data Structures in Bioinformatics
dsb-meeting.github.io
December 10, 2025 at 2:29 PM
The 12th edition of the 2-days workshop “Data Structures in Bioinformatics” (DSB) will take place in Venice (Italy) on February 18-19th, 2026: dsb-meeting.github.io/DSB2026/
Reposted by Roland Faure

1/9 Just out:
k-mer indexes are the backbone of fast search in genomic data, but many degrade under small k, subsampling, or high diversity.
With Ondřej Sladký and @pavelvesely.bsky.social we asked: can we build one that works efficiently for any k-mer set?
k-mer indexes are the backbone of fast search in genomic data, but many degrade under small k, subsampling, or high diversity.
With Ondřej Sladký and @pavelvesely.bsky.social we asked: can we build one that works efficiently for any k-mer set?
🧮 Just out in Bioinformatics Advances: “FroM Superstring to Indexing: A space-efficient index for unconstrained k-mer sets using the Masked Burrows-Wheeler Transform (MBWT)”
Full article available: https://doi.org/10.1093/bioadv/vbaf290
Authors include: @pavelvesely.bsky.social, @brinda.eu
Full article available: https://doi.org/10.1093/bioadv/vbaf290
Authors include: @pavelvesely.bsky.social, @brinda.eu

December 5, 2025 at 5:42 PM
1/9 Just out:
k-mer indexes are the backbone of fast search in genomic data, but many degrade under small k, subsampling, or high diversity.
With Ondřej Sladký and @pavelvesely.bsky.social we asked: can we build one that works efficiently for any k-mer set?
k-mer indexes are the backbone of fast search in genomic data, but many degrade under small k, subsampling, or high diversity.
With Ondřej Sladký and @pavelvesely.bsky.social we asked: can we build one that works efficiently for any k-mer set?
Preprint out! Check out our new long-read metagenomic SNP-caller, SNooPy 😀. Work with Chris Quince. Thread 🧵
👉 www.biorxiv.org/content/10.6...
👉 www.biorxiv.org/content/10.6...

December 4, 2025 at 1:18 PM
Preprint out! Check out our new long-read metagenomic SNP-caller, SNooPy 😀. Work with Chris Quince. Thread 🧵
👉 www.biorxiv.org/content/10.6...
👉 www.biorxiv.org/content/10.6...
Reposted by Roland Faure

Preprint Alert!
We present new strategies to accelerate large-scale document comparison using MinHash-like sketches.
A thread:
We present new strategies to accelerate large-scale document comparison using MinHash-like sketches.
A thread:
Compressed inverted indexes for scalable sequence similarity https://www.biorxiv.org/content/10.1101/2025.11.21.689685v1
December 1, 2025 at 2:59 PM
Preprint Alert!
We present new strategies to accelerate large-scale document comparison using MinHash-like sketches.
A thread:
We present new strategies to accelerate large-scale document comparison using MinHash-like sketches.
A thread:
Our preprint on our new metagenomic HiFi assembler Alice is out 🥳 Based on a *new sketching method* (🧵1/6)
👉 Preprint www.biorxiv.org/content/10.1...
👉 Github github.com/rolandfaure/...
👉 Preprint www.biorxiv.org/content/10.1...
👉 Github github.com/rolandfaure/...

Alice: fast and haplotype-aware assembly of high-fidelity reads based on MSR sketching
We introduce Mapping-friendly Sequence Reduction (MSR) sketches, a sketching method for high-fidelity (HiFi) long reads, and Alice, an assembler that operates directly on these sketches. MSR produces ...
www.biorxiv.org
October 3, 2025 at 2:51 PM
Our preprint on our new metagenomic HiFi assembler Alice is out 🥳 Based on a *new sketching method* (🧵1/6)
👉 Preprint www.biorxiv.org/content/10.1...
👉 Github github.com/rolandfaure/...
👉 Preprint www.biorxiv.org/content/10.1...
👉 Github github.com/rolandfaure/...
Reposted by Roland Faure

🌎👩🔬 For 15+ years biology has accumulated petabytes (million gigabytes) of🧬DNA sequencing data🧬 from the far reaches of our planet.🦠🍄🌵
Logan now democratizes efficient access to the world’s most comprehensive genetics dataset. Free and open.
doi.org/10.1101/2024...
Logan now democratizes efficient access to the world’s most comprehensive genetics dataset. Free and open.
doi.org/10.1101/2024...

September 3, 2025 at 8:39 AM
🌎👩🔬 For 15+ years biology has accumulated petabytes (million gigabytes) of🧬DNA sequencing data🧬 from the far reaches of our planet.🦠🍄🌵
Logan now democratizes efficient access to the world’s most comprehensive genetics dataset. Free and open.
doi.org/10.1101/2024...
Logan now democratizes efficient access to the world’s most comprehensive genetics dataset. Free and open.
doi.org/10.1101/2024...
Reposted by Roland Faure

Preprint out for myloasm, our new nanopore / HiFi metagenome assembler!
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
High-resolution metagenome assembly for modern long reads with myloasm https://www.biorxiv.org/content/10.1101/2025.09.05.674543v1
September 7, 2025 at 11:35 PM
Preprint out for myloasm, our new nanopore / HiFi metagenome assembler!
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
Reposted by Roland Faure

I am happy to share our new preprint introducing MADRe - a pipeline for Metagenomic Assembly-Driven Database Reduction, enabling accurate and computationally efficient strain-level metagenomic classification.
🔗https://www.biorxiv.org/content/10.1101/2025.05.12.653324v1
1/9
🔗https://www.biorxiv.org/content/10.1101/2025.05.12.653324v1
1/9
May 16, 2025 at 8:37 AM
I am happy to share our new preprint introducing MADRe - a pipeline for Metagenomic Assembly-Driven Database Reduction, enabling accurate and computationally efficient strain-level metagenomic classification.
🔗https://www.biorxiv.org/content/10.1101/2025.05.12.653324v1
1/9
🔗https://www.biorxiv.org/content/10.1101/2025.05.12.653324v1
1/9
Reposted by Roland Faure

Starting #RECOMBseq with @rayanchikhi.bsky.social 's keynote. Here stressing our responsibility as scientists to enable access to a common good: genomic data

April 24, 2025 at 12:42 AM
Starting #RECOMBseq with @rayanchikhi.bsky.social 's keynote. Here stressing our responsibility as scientists to enable access to a common good: genomic data
Reposted by Roland Faure

Side note: you could, speaking purely theoretically, also fit every microbe onto an SD card, which is within the weight limit for a carrier pigeon. For some distances, it would be faster than the internet for transmitting sequence libraries
7/
7/

April 9, 2025 at 9:10 PM
Side note: you could, speaking purely theoretically, also fit every microbe onto an SD card, which is within the weight limit for a carrier pigeon. For some distances, it would be faster than the internet for transmitting sequence libraries
7/
7/
Reposted by Roland Faure
So glad this is finally out. The method has been instrumental in allowing us to compress the AllTheBacteria data - ~2 million bacterial genomes shrink from 3Terabytes (gzipped) to 100Gb using phylogenetic compression. Great work by @brinda.eu
Our latest paper, in which @brinda.eu (along with @zaminiqbal.bsky.social and others) introduces phylogenetic compression for storage and search of enormous microbial genome libraries, was published today in @naturemethods.bsky.social:
rdcu.be/eg4OA
1/
rdcu.be/eg4OA
1/
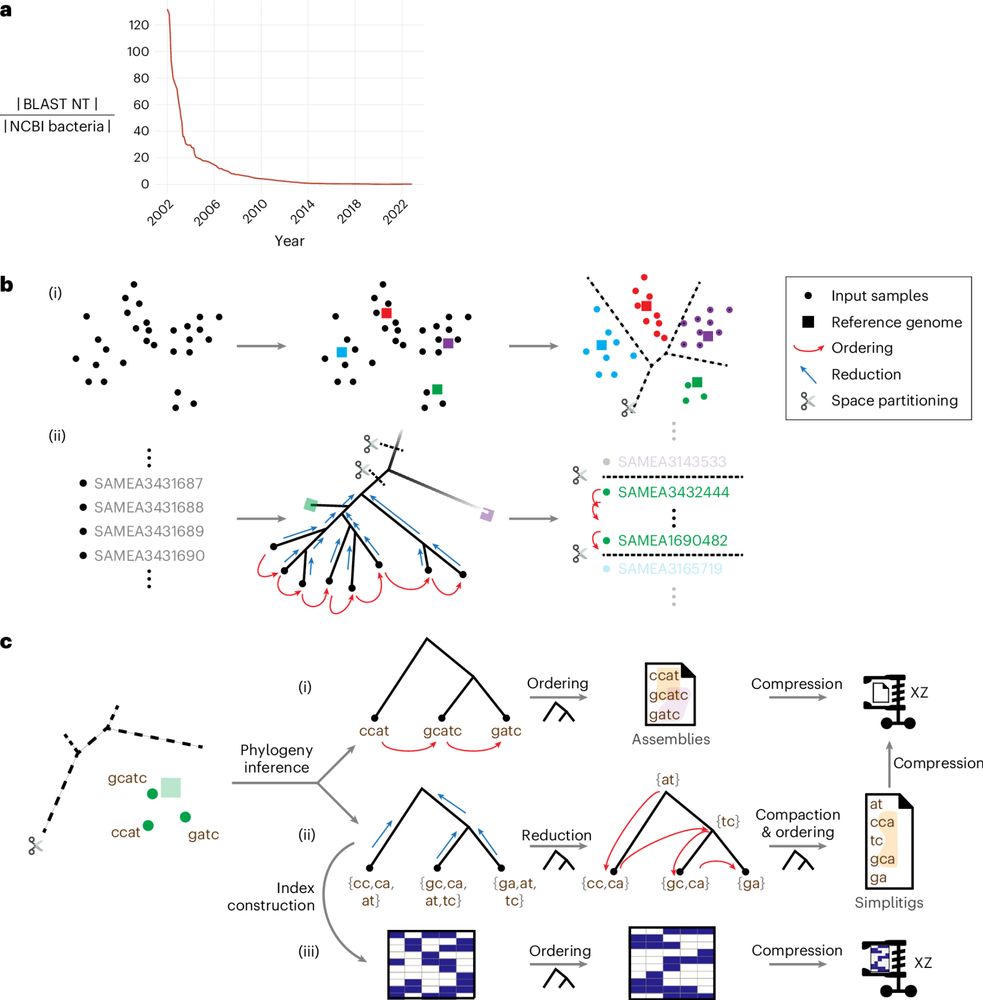
Efficient and robust search of microbial genomes via phylogenetic compression
Nature Methods - Phylogenetic compression achieves performant and lossless compression of massive collections of microbial genomes, facilitating fast BLAST-like search and versatile alignment tasks.
rdcu.be
April 9, 2025 at 10:27 PM
So glad this is finally out. The method has been instrumental in allowing us to compress the AllTheBacteria data - ~2 million bacterial genomes shrink from 3Terabytes (gzipped) to 100Gb using phylogenetic compression. Great work by @brinda.eu
Reposted by Roland Faure

Do you (like me) create a bunch of conda environments, then later forget what they're for, when they were last updated, or which tools are in them?
If so, you might this little project: github.com/rrwick/conda...
If so, you might this little project: github.com/rrwick/conda...
GitHub - rrwick/condaenvlist: a simple tool for listing conda environments with descriptions
a simple tool for listing conda environments with descriptions - rrwick/condaenvlist
github.com
March 27, 2025 at 4:34 AM
Do you (like me) create a bunch of conda environments, then later forget what they're for, when they were last updated, or which tools are in them?
If so, you might this little project: github.com/rrwick/conda...
If so, you might this little project: github.com/rrwick/conda...
So glad to have participated in #DSB2025, what a great workshop! For some mysterious reason it was the first time I attended after 3 years of sequence research. Thanks to all participants & organizers 😃

March 7, 2025 at 7:41 PM
So glad to have participated in #DSB2025, what a great workshop! For some mysterious reason it was the first time I attended after 3 years of sequence research. Thanks to all participants & organizers 😃
Reposted by Roland Faure

Ragnar's made some incredible optimizations on the computation of minimizers, can't wait to see how these improvements will benefit bioinfo tools!

Nice result to end the day (night*):
After discussions with @imartayan.bsky.social, the SIMD minimizer code now also does proper canonical (revcomp) minimizers:
~1ns/bp for fwd minis
+0.4ns/bp with collect and dedup
+0.6ns/bp with canonical hashes.
Super happy how it's only 2x slower in the end!
After discussions with @imartayan.bsky.social, the SIMD minimizer code now also does proper canonical (revcomp) minimizers:
~1ns/bp for fwd minis
+0.4ns/bp with collect and dedup
+0.6ns/bp with canonical hashes.
Super happy how it's only 2x slower in the end!
December 13, 2024 at 3:50 PM
Ragnar's made some incredible optimizations on the computation of minimizers, can't wait to see how these improvements will benefit bioinfo tools!
Reposted by Roland Faure

Amazing ideas here www.biorxiv.org/content/bior... from
@yoann.bsky.social
and collaborators.
Reorganize minimizers to allow kmers dichotomic search. That's brilliant.
#bioinformatics 🧬🖥️
@yoann.bsky.social
and collaborators.
Reorganize minimizers to allow kmers dichotomic search. That's brilliant.
#bioinformatics 🧬🖥️

December 4, 2024 at 12:10 PM
Amazing ideas here www.biorxiv.org/content/bior... from
@yoann.bsky.social
and collaborators.
Reorganize minimizers to allow kmers dichotomic search. That's brilliant.
#bioinformatics 🧬🖥️
@yoann.bsky.social
and collaborators.
Reorganize minimizers to allow kmers dichotomic search. That's brilliant.
#bioinformatics 🧬🖥️
So glad to have successfully defended my Ph.D. last week 😀 Work on producing haplotype-resolved metagenomic assemblies using noisy long reads (HairSplitter) and high-fidelity long reads (Alice assembler, unpublished yet).
Thanks to my advisors Dominique Lavenier and Jean-François Flot ❤️
Thanks to my advisors Dominique Lavenier and Jean-François Flot ❤️

December 4, 2024 at 8:32 AM
So glad to have successfully defended my Ph.D. last week 😀 Work on producing haplotype-resolved metagenomic assemblies using noisy long reads (HairSplitter) and high-fidelity long reads (Alice assembler, unpublished yet).
Thanks to my advisors Dominique Lavenier and Jean-François Flot ❤️
Thanks to my advisors Dominique Lavenier and Jean-François Flot ❤️