




Gu Zhenhao
@guzhenhao.bsky.social
PhD student at NUS Computing, interested in algorithms for Computational Biology.
Reposted by Gu Zhenhao

Really exciting that the preprint on Barbell, a new demultiplexer, is finally out!
It's the first tool that builds on Sassy, the approximate-DNA-searching tool that @rickbitloo.bsky.social and myself developed earlier this year, specifically with this application in mind.
It's the first tool that builds on Sassy, the approximate-DNA-searching tool that @rickbitloo.bsky.social and myself developed earlier this year, specifically with this application in mind.

Around 10% of your Nanopore reads (SQK-RBK114) are incorrectly trimmed. Here is why, and how our new tool Barbell solves it:
www.biorxiv.org/content/10.1...
Want to get started? github.com/rickbeeloo/b...
www.biorxiv.org/content/10.1...
Want to get started? github.com/rickbeeloo/b...
October 23, 2025 at 9:28 PM
Really exciting that the preprint on Barbell, a new demultiplexer, is finally out!
It's the first tool that builds on Sassy, the approximate-DNA-searching tool that @rickbitloo.bsky.social and myself developed earlier this year, specifically with this application in mind.
It's the first tool that builds on Sassy, the approximate-DNA-searching tool that @rickbitloo.bsky.social and myself developed earlier this year, specifically with this application in mind.
Reposted by Gu Zhenhao

1/6 Movi 2 is here: faster and more space-efficient for pangenome queries. Its fastest mode uses half the memory of Movi 1 while running ~30% faster. github.com/mohsenzakeri...

GitHub - mohsenzakeri/Movi: Fast, Cache-Efficient, and Scalable Queries on Pangenomes
Fast, Cache-Efficient, and Scalable Queries on Pangenomes - mohsenzakeri/Movi
github.com
October 21, 2025 at 8:00 PM
1/6 Movi 2 is here: faster and more space-efficient for pangenome queries. Its fastest mode uses half the memory of Movi 1 while running ~30% faster. github.com/mohsenzakeri...
Reposted by Gu Zhenhao

I'm excited to share our pre-print about a new variant benchmarking tool we've been working on for the past few months!
Aardvark: Sifting through differences in a mound of variants
GitHub: github.com/PacificBiosc...
Some highlights in this thread:
1/N
Aardvark: Sifting through differences in a mound of variants
GitHub: github.com/PacificBiosc...
Some highlights in this thread:
1/N
October 6, 2025 at 8:07 PM
I'm excited to share our pre-print about a new variant benchmarking tool we've been working on for the past few months!
Aardvark: Sifting through differences in a mound of variants
GitHub: github.com/PacificBiosc...
Some highlights in this thread:
1/N
Aardvark: Sifting through differences in a mound of variants
GitHub: github.com/PacificBiosc...
Some highlights in this thread:
1/N
Reposted by Gu Zhenhao

Our new tool "X-Mapper: fast and accurate sequence alignment via gapped x-mers" now published on Genome Biology! Please try it if you work on DNA sequences :) github.com/mathjeff/Map...
genomebiology.biomedcentral.com/articles/10....
genomebiology.biomedcentral.com/articles/10....
X-Mapper: fast and accurate sequence alignment via gapped x-mers - Genome Biology
Sequence alignment is foundational to many bioinformatic analyses. Many aligners start by splitting sequences into contiguous, fixed-length seeds, called k-mers. Alignment is faster with longer, uniqu...
genomebiology.biomedcentral.com
August 30, 2025 at 1:45 AM
Our new tool "X-Mapper: fast and accurate sequence alignment via gapped x-mers" now published on Genome Biology! Please try it if you work on DNA sequences :) github.com/mathjeff/Map...
genomebiology.biomedcentral.com/articles/10....
genomebiology.biomedcentral.com/articles/10....
Reposted by Gu Zhenhao

🦒Long read giraffe is out!🦒
Mapping long reads to pangenome graphs is ~10x faster than with GraphAligner, with veeery slightly better mapping accuracy, short variant calling, and SV genotyping than GraphAligner or Minimap2
Mapping long reads to pangenome graphs is ~10x faster than with GraphAligner, with veeery slightly better mapping accuracy, short variant calling, and SV genotyping than GraphAligner or Minimap2
Rapid, accurate long- and short-read mapping to large pangenome graphs with vg Giraffe https://www.biorxiv.org/content/10.1101/2025.09.29.678807v1
October 2, 2025 at 6:28 AM
🦒Long read giraffe is out!🦒
Mapping long reads to pangenome graphs is ~10x faster than with GraphAligner, with veeery slightly better mapping accuracy, short variant calling, and SV genotyping than GraphAligner or Minimap2
Mapping long reads to pangenome graphs is ~10x faster than with GraphAligner, with veeery slightly better mapping accuracy, short variant calling, and SV genotyping than GraphAligner or Minimap2
Reposted by Gu Zhenhao

Precisely calling mutations across hundreds of bacterial isolates has been hard, requiring manual filtering and expertise.
Until now, using AccuSNV.
Herui Liao trained an ML model based on our previous meticulously called SNVs.
www.biorxiv.org/content/10.1...
Until now, using AccuSNV.
Herui Liao trained an ML model based on our previous meticulously called SNVs.
www.biorxiv.org/content/10.1...

High-accuracy SNV calling for bacterial isolates using deep learning with AccuSNV
Accurate detection of mutations within bacterial species is critical for fundamental studies of microbial evolution, reconstructing transmission events, and identifying antimicrobial resistance mutati...
www.biorxiv.org
September 29, 2025 at 7:45 PM
Precisely calling mutations across hundreds of bacterial isolates has been hard, requiring manual filtering and expertise.
Until now, using AccuSNV.
Herui Liao trained an ML model based on our previous meticulously called SNVs.
www.biorxiv.org/content/10.1...
Until now, using AccuSNV.
Herui Liao trained an ML model based on our previous meticulously called SNVs.
www.biorxiv.org/content/10.1...


Reposted by Gu Zhenhao

Excited to share our EvANI benchmarking workflow, published in Briefings in Bioinformatics doi.org/10.1093/bib/...
Computing average nucleotide identity (ANI) is neither conceptually nor computationally trivial. Its definition has evolved over years, with different meanings and assumptions (1/5)
Computing average nucleotide identity (ANI) is neither conceptually nor computationally trivial. Its definition has evolved over years, with different meanings and assumptions (1/5)

September 21, 2025 at 3:26 PM
Excited to share our EvANI benchmarking workflow, published in Briefings in Bioinformatics doi.org/10.1093/bib/...
Computing average nucleotide identity (ANI) is neither conceptually nor computationally trivial. Its definition has evolved over years, with different meanings and assumptions (1/5)
Computing average nucleotide identity (ANI) is neither conceptually nor computationally trivial. Its definition has evolved over years, with different meanings and assumptions (1/5)
Reposted by Gu Zhenhao

Excited to share our latest preprint on agtools, an open-source Python framework for analysing and manipulating assembly graphs. (1/n)
www.biorxiv.org/content/10.1...
#Bioinformatics #genomics #assembly #assemblygraphs #software
www.biorxiv.org/content/10.1...
#Bioinformatics #genomics #assembly #assemblygraphs #software

agtools: a software framework to manipulate assembly graphs
Assembly graphs are a fundamental data structure used by genome and metagenome assemblers to represent sequences and their overlap information, facilitating the assembler to construct longer genomic f...
www.biorxiv.org
September 17, 2025 at 6:58 AM
Excited to share our latest preprint on agtools, an open-source Python framework for analysing and manipulating assembly graphs. (1/n)
www.biorxiv.org/content/10.1...
#Bioinformatics #genomics #assembly #assemblygraphs #software
www.biorxiv.org/content/10.1...
#Bioinformatics #genomics #assembly #assemblygraphs #software
Reposted by Gu Zhenhao
New blog post – A quick look at Roche's SBX
lh3.github.io/2025/09/11/a...
lh3.github.io/2025/09/11/a...

September 12, 2025 at 3:26 AM
New blog post – A quick look at Roche's SBX
lh3.github.io/2025/09/11/a...
lh3.github.io/2025/09/11/a...
Reposted by Gu Zhenhao

Sometimes you meet absolutely incredible bioinfo-magicians.
It was a huge privilege when @shenwei356.bsky.social
joined our group for a year on an @embl.org sabbatical.
While here, he developed a new way of aligning to
millions of bacteria, called LexicMap 1/n
www.nature.com/articles/s41...
It was a huge privilege when @shenwei356.bsky.social
joined our group for a year on an @embl.org sabbatical.
While here, he developed a new way of aligning to
millions of bacteria, called LexicMap 1/n
www.nature.com/articles/s41...

Efficient sequence alignment against millions of prokaryotic genomes with LexicMap - Nature Biotechnology
LexicMap uses a fixed set of probes to efficiently query gene sequences for fast and low-memory alignment.
www.nature.com
September 10, 2025 at 9:12 AM
Sometimes you meet absolutely incredible bioinfo-magicians.
It was a huge privilege when @shenwei356.bsky.social
joined our group for a year on an @embl.org sabbatical.
While here, he developed a new way of aligning to
millions of bacteria, called LexicMap 1/n
www.nature.com/articles/s41...
It was a huge privilege when @shenwei356.bsky.social
joined our group for a year on an @embl.org sabbatical.
While here, he developed a new way of aligning to
millions of bacteria, called LexicMap 1/n
www.nature.com/articles/s41...
Reposted by Gu Zhenhao

Preprint out for myloasm, our new nanopore / HiFi metagenome assembler!
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
High-resolution metagenome assembly for modern long reads with myloasm https://www.biorxiv.org/content/10.1101/2025.09.05.674543v1
September 7, 2025 at 11:35 PM
Preprint out for myloasm, our new nanopore / HiFi metagenome assembler!
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
Reposted by Gu Zhenhao

Published finally! Our study describing the use of metagenomics for AMR/pathogen surveillance in food-centres island-wide in Singapore is out in npj Antimicrobial & Resistance:
nature.com/articles/s44...
nature.com/articles/s44...

Citywide metagenomic surveillance of food centres reveals local microbial signatures and antibiotic resistance gene enrichment - npj Antimicrobials and Resistance
npj Antimicrobials and Resistance - Citywide metagenomic surveillance of food centres reveals local microbial signatures and antibiotic resistance gene enrichment
nature.com
September 6, 2025 at 2:31 AM
Published finally! Our study describing the use of metagenomics for AMR/pathogen surveillance in food-centres island-wide in Singapore is out in npj Antimicrobial & Resistance:
nature.com/articles/s44...
nature.com/articles/s44...
Reposted by Gu Zhenhao

High-resolution metagenome assembly for modern long reads with myloasm https://www.biorxiv.org/content/10.1101/2025.09.05.674543v1
September 7, 2025 at 4:47 AM
High-resolution metagenome assembly for modern long reads with myloasm https://www.biorxiv.org/content/10.1101/2025.09.05.674543v1
Reposted by Gu Zhenhao
Our study developing a skin metatranscriptomics protocol is now out in @natbiotech.nature.com!
We finally have the ability to study microbial activity on skin and identify key functional genes playing a role in diseases.
Amazing team of Chia Minghao and Amanda Ng 👏
nature.com/articles/s41...
We finally have the ability to study microbial activity on skin and identify key functional genes playing a role in diseases.
Amazing team of Chia Minghao and Amanda Ng 👏
nature.com/articles/s41...
August 30, 2025 at 2:17 AM
Our study developing a skin metatranscriptomics protocol is now out in @natbiotech.nature.com!
We finally have the ability to study microbial activity on skin and identify key functional genes playing a role in diseases.
Amazing team of Chia Minghao and Amanda Ng 👏
nature.com/articles/s41...
We finally have the ability to study microbial activity on skin and identify key functional genes playing a role in diseases.
Amazing team of Chia Minghao and Amanda Ng 👏
nature.com/articles/s41...
Reposted by Gu Zhenhao

Wow, more than 2.4M of assembled bacteria in the new release of ABT! We plan to index these using our efficient colored De Bruijn graph index, Fulgor. We recently conducted experiments with nearly 1M genomes…getting there :)
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...

AllTheBacteria - all bacterial genomes assembled, available and searchable
The bacterial sequence data publicly available via the global DNA archives is a vast potential source of information on the evolution of bacteria. However, most of this sequence data is unassembled, o...
www.biorxiv.org
August 28, 2025 at 12:16 PM
Wow, more than 2.4M of assembled bacteria in the new release of ABT! We plan to index these using our efficient colored De Bruijn graph index, Fulgor. We recently conducted experiments with nearly 1M genomes…getting there :)
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
Reposted by Gu Zhenhao

The microbial composition of metagenomes is identified in seconds by profiling against large databases go.nature.com/3BBVqDC
rdcu.be/eCbj4
rdcu.be/eCbj4
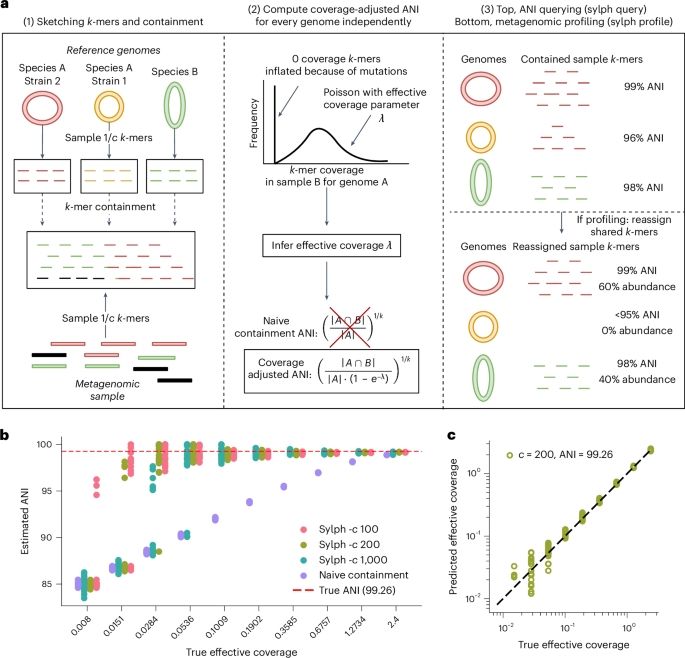
Rapid species-level metagenome profiling and containment estimation with sylph - Nature Biotechnology
The microbial composition of metagenomes is identified in seconds by profiling against large databases.
go.nature.com
August 27, 2025 at 1:14 AM
The microbial composition of metagenomes is identified in seconds by profiling against large databases go.nature.com/3BBVqDC
rdcu.be/eCbj4
rdcu.be/eCbj4
Reposted by Gu Zhenhao
Our work on direct @nanoporetech.com sequencing of non-canonical bases in now out in @natcomms.nature.com!
Read all about it here: nature.com/articles/s41...
Great collab with Chew and Hirao lab
x.com/NiranjanTW/s...
Read all about it here: nature.com/articles/s41...
Great collab with Chew and Hirao lab
x.com/NiranjanTW/s...

Direct high-throughput deconvolution of non-canonical bases via nanopore sequencing and bootstrapped learning - Nature Communications
Perez, Kimoto, Rajakumar and colleagues present a fast and accurate DNA sequencing method that reads canonical and non-canonical bases using AI and nanopore technology. The approach enables an expande...
nature.com
August 22, 2025 at 9:32 AM
Our work on direct @nanoporetech.com sequencing of non-canonical bases in now out in @natcomms.nature.com!
Read all about it here: nature.com/articles/s41...
Great collab with Chew and Hirao lab
x.com/NiranjanTW/s...
Read all about it here: nature.com/articles/s41...
Great collab with Chew and Hirao lab
x.com/NiranjanTW/s...
Reposted by Gu Zhenhao
Great talk by Vikram @vikramshivakumar.bsky.social on studying pangenomes and synteny visualization in #WABI25
Github: github.com/vikshiv/mume...
First paper: genomebiology.biomedcentral.com/articles/10....
Second: www.biorxiv.org/content/10.1... #WABI2025
Github: github.com/vikshiv/mume...
First paper: genomebiology.biomedcentral.com/articles/10....
Second: www.biorxiv.org/content/10.1... #WABI2025



August 20, 2025 at 3:03 PM
Great talk by Vikram @vikramshivakumar.bsky.social on studying pangenomes and synteny visualization in #WABI25
Github: github.com/vikshiv/mume...
First paper: genomebiology.biomedcentral.com/articles/10....
Second: www.biorxiv.org/content/10.1... #WABI2025
Github: github.com/vikshiv/mume...
First paper: genomebiology.biomedcentral.com/articles/10....
Second: www.biorxiv.org/content/10.1... #WABI2025
Reposted by Gu Zhenhao

Manually curated and harmonized metadata for over 110k metagenomic samples! (58k samples from the human gut alone!)🦠
Proud to have contributed to Metalog, the latest @borklab.bsky.social resource:
www.biorxiv.org/content/10.1...
#microsky #microbiome
Proud to have contributed to Metalog, the latest @borklab.bsky.social resource:
www.biorxiv.org/content/10.1...
#microsky #microbiome
August 15, 2025 at 1:54 PM
Manually curated and harmonized metadata for over 110k metagenomic samples! (58k samples from the human gut alone!)🦠
Proud to have contributed to Metalog, the latest @borklab.bsky.social resource:
www.biorxiv.org/content/10.1...
#microsky #microbiome
Proud to have contributed to Metalog, the latest @borklab.bsky.social resource:
www.biorxiv.org/content/10.1...
#microsky #microbiome
Reposted by Gu Zhenhao

Interested in a tool that aligns millions of proteins in minutes with quality similar to or better than the state-of-the-art utilities? Please take a look at our FAMSA2 paper: www.biorxiv.org/content/10.1...
and GH repo: github.com/refresh-bio/...
and GH repo: github.com/refresh-bio/...

FAMSA2 enables accurate multiple sequence alignment at protein-universe scale
We introduce FAMSA2, an algorithm that produces high-accuracy multiple protein sequence alignments with unprecedented speed. Across structural, phylogenetic, and functional benchmarks, FAMSA2 matches ...
www.biorxiv.org
July 19, 2025 at 9:28 PM
Interested in a tool that aligns millions of proteins in minutes with quality similar to or better than the state-of-the-art utilities? Please take a look at our FAMSA2 paper: www.biorxiv.org/content/10.1...
and GH repo: github.com/refresh-bio/...
and GH repo: github.com/refresh-bio/...
Reposted by Gu Zhenhao

Out in @natbiotech.nature.com: Metagenome taxonomy profilers usually ignore unknown species. SingleM is an accurate profiler which doesn't, even detecting phyla with no MAGs. Profiles of 700,000 metagenomes at sandpiper.qut.edu.au. A 🧵

July 16, 2025 at 9:59 PM
Out in @natbiotech.nature.com: Metagenome taxonomy profilers usually ignore unknown species. SingleM is an accurate profiler which doesn't, even detecting phyla with no MAGs. Profiles of 700,000 metagenomes at sandpiper.qut.edu.au. A 🧵
Reposted by Gu Zhenhao
Sassy is out now!
Ever need to search for approximate matches of short DNA strings?
Sassy is the tool to use!
Available now wherever you get your code
With @rickbitloo.bsky.social
curiouscoding.nl/papers/sassy...
github.com/ragnarGrootK...
Ever need to search for approximate matches of short DNA strings?
Sassy is the tool to use!
Available now wherever you get your code
With @rickbitloo.bsky.social
curiouscoding.nl/papers/sassy...
github.com/ragnarGrootK...
July 18, 2025 at 8:20 PM
Sassy is out now!
Ever need to search for approximate matches of short DNA strings?
Sassy is the tool to use!
Available now wherever you get your code
With @rickbitloo.bsky.social
curiouscoding.nl/papers/sassy...
github.com/ragnarGrootK...
Ever need to search for approximate matches of short DNA strings?
Sassy is the tool to use!
Available now wherever you get your code
With @rickbitloo.bsky.social
curiouscoding.nl/papers/sassy...
github.com/ragnarGrootK...
Reposted by Gu Zhenhao

The GreedyMini paper is finally out as part of #ISMBECCB2025 proceedings: academic.oup.com/bioinformati.... Arseny will present it on the last day (July 24th) and last session (14:20) of the HiTSeq meeting.
GreedyMini: generating low-density DNA minimizers
AbstractMotivation. Minimizers are the most popular k-mer selection scheme in algorithms and data structures analyzing high-throughput sequencing (HTS) dat
academic.oup.com
July 16, 2025 at 6:31 AM
The GreedyMini paper is finally out as part of #ISMBECCB2025 proceedings: academic.oup.com/bioinformati.... Arseny will present it on the last day (July 24th) and last session (14:20) of the HiTSeq meeting.
Reposted by Gu Zhenhao

Maxime Crochemore, Thierry Lecroq, Wojtek Rytter
25 Additional Problems -- Extension to the Book "125 Problems in Text Algorithms"
https://arxiv.org/abs/2507.05770
25 Additional Problems -- Extension to the Book "125 Problems in Text Algorithms"
https://arxiv.org/abs/2507.05770
July 9, 2025 at 4:07 AM
Maxime Crochemore, Thierry Lecroq, Wojtek Rytter
25 Additional Problems -- Extension to the Book "125 Problems in Text Algorithms"
https://arxiv.org/abs/2507.05770
25 Additional Problems -- Extension to the Book "125 Problems in Text Algorithms"
https://arxiv.org/abs/2507.05770