



Xi Fu
@fuxialexander.bsky.social
Transcription regulation; deep learning; (bad) developer
Reposted by Xi Fu

New work from the lab trying to wrap our heads around the massive complexity of the human transcriptome revealed by long-read RNA-seq! Fun collab with Gloria Sheynkman. www.biorxiv.org/content/10.1...

Perplexity as a Metric for Isoform Diversity in the Human Transcriptome
Long-read sequencing (LRS) has revealed a far greater diversity of RNA isoforms than earlier technologies, increasing the critical need to determine which, and how many, isoforms per gene are biologic...
www.biorxiv.org
July 2, 2025 at 11:46 PM
New work from the lab trying to wrap our heads around the massive complexity of the human transcriptome revealed by long-read RNA-seq! Fun collab with Gloria Sheynkman. www.biorxiv.org/content/10.1...
Reposted by Xi Fu

We have updated our protein lanuage model trained on structure dynamics. Our new models show significant better zero-shot performance on mutation effects of designed and viral proteins compared to ESM2. check the new preprint here: www.biorxiv.org/content/10.1...
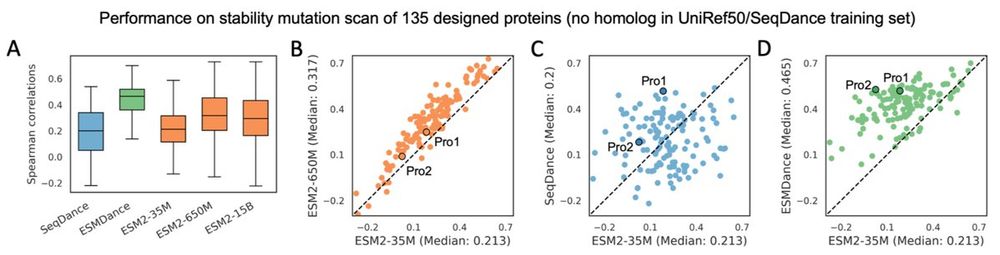
April 17, 2025 at 2:40 PM
We have updated our protein lanuage model trained on structure dynamics. Our new models show significant better zero-shot performance on mutation effects of designed and viral proteins compared to ESM2. check the new preprint here: www.biorxiv.org/content/10.1...
Reposted by Xi Fu

Some encouraging news for cross-gene generalization of allele effects in S2F models. www.biorxiv.org/content/10.1...

Deep genomic models of allele-specific measurements
Allele-specific quantification of sequencing data, such as gene expression, allows for a causal investigation of how DNA sequence variations influence cis gene regulation. Current methods for analyzin...
www.biorxiv.org
April 16, 2025 at 1:46 AM
Some encouraging news for cross-gene generalization of allele effects in S2F models. www.biorxiv.org/content/10.1...
Reposted by Xi Fu

An engineered Cas12a enables higher-order combinatorial functional genomic screens using CRISPR interference go.nature.com/3UTnSXM
rdcu.be/ef95k
rdcu.be/ef95k

Engineered CRISPR-Cas12a for higher-order combinatorial chromatin perturbations - Nature Biotechnology
An engineered Cas12a enables higher-order combinatorial functional genomic screens using CRISPR interference.
go.nature.com
April 3, 2025 at 2:30 AM
An engineered Cas12a enables higher-order combinatorial functional genomic screens using CRISPR interference go.nature.com/3UTnSXM
rdcu.be/ef95k
rdcu.be/ef95k
Reposted by Xi Fu

1/10 Excited to share our latest - the first whole-body map of both DNA methylation and 3D genome at single-cell resolution.
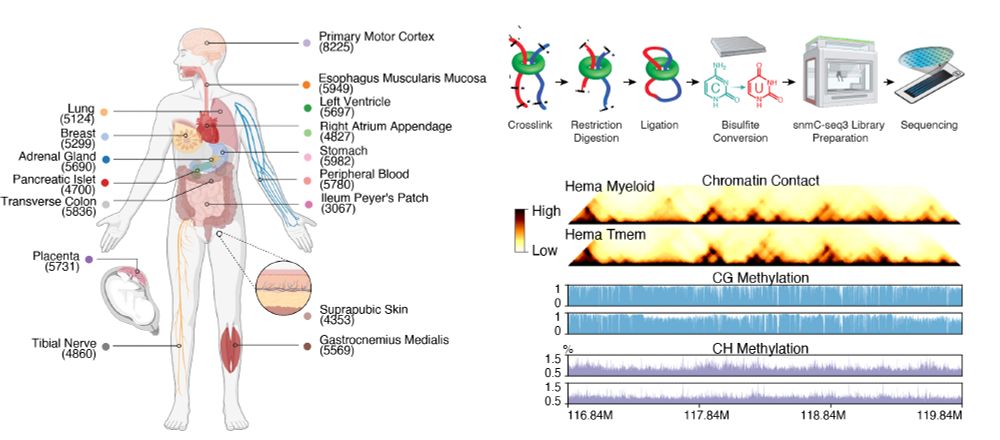
March 25, 2025 at 3:49 PM
1/10 Excited to share our latest - the first whole-body map of both DNA methylation and 3D genome at single-cell resolution.
Biorxiv seems to be really slow nowadays. Is it just me? Curious whether it's due to some infra change or there are some AI Agents crawling the data...
March 21, 2025 at 3:16 PM
Biorxiv seems to be really slow nowadays. Is it just me? Curious whether it's due to some infra change or there are some AI Agents crawling the data...
Reposted by Xi Fu
For decades, government funding “has positioned the United States as a global leader” in science, says scientist Tom Maniatis of @zuckermanbrain.bsky.social and the New York Genome Center. He highlights how a new #NIH policy cutting money for research “jeopardizes” this, in Cell tinyurl.com/ubw6uphe

March 17, 2025 at 5:04 PM
For decades, government funding “has positioned the United States as a global leader” in science, says scientist Tom Maniatis of @zuckermanbrain.bsky.social and the New York Genome Center. He highlights how a new #NIH policy cutting money for research “jeopardizes” this, in Cell tinyurl.com/ubw6uphe
Reposted by Xi Fu

Can someone send this to the NIH Director nominee who said yesterday under oath that he doesn’t know where the indirects go.
March 6, 2025 at 5:59 PM
Can someone send this to the NIH Director nominee who said yesterday under oath that he doesn’t know where the indirects go.
Reposted by Xi Fu

We are crowd sourcing reductions in graduate admissions and hiring freezes across biomedical research and higher ed in response to pauses in NIH funding and EO’s. If you have information if you could add to this spreadsheet, it would be greatly appreciated!: docs.google.com/spreadsheets...
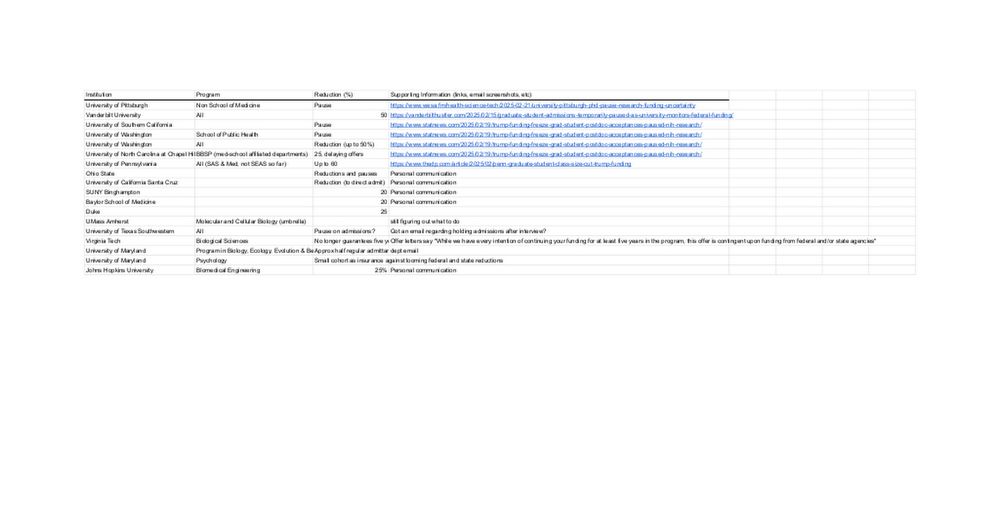
Graduate Reductions Across Biomedical Sciences (2025)
docs.google.com
February 22, 2025 at 6:09 PM
We are crowd sourcing reductions in graduate admissions and hiring freezes across biomedical research and higher ed in response to pauses in NIH funding and EO’s. If you have information if you could add to this spreadsheet, it would be greatly appreciated!: docs.google.com/spreadsheets...
Reposted by Xi Fu
This is very cool work (where I was fortunate to play a small part), providing creative and crucial solutions for secure and federated eQTL mapping. Bigger functional genetic studies with less administrative and legal hassle! 💪

The world is on fire but we must continue doing science! With immense pride, I share the latest from my lab: privateQTL - a method for federated and secure eQTL mapping, led by my brilliant student Annie from @ColumbiaDBMI www.cell.com/cell-genomic...
(1/n)
(1/n)

Secure and federated quantitative trait loci mapping with privateQTL
Choi et al. developed a novel tool for privacy-preserving cross-institutional eQTL
mapping studies. The authors benchmarked their tool against meta-analysis and demonstrated
that it achieves higher ac...
www.cell.com
February 12, 2025 at 4:34 PM
This is very cool work (where I was fortunate to play a small part), providing creative and crucial solutions for secure and federated eQTL mapping. Bigger functional genetic studies with less administrative and legal hassle! 💪
Reposted by Xi Fu

Combinatorial mapping of E3 ubiquitin ligases to their target substrates
www.cell.com/molecular-ce...
www.cell.com/molecular-ce...

Combinatorial mapping of E3 ubiquitin ligases to their target substrates
The substrate(s) of most E3 ubiquitin ligases remain unknown. Suiter et al. present
COMET, a combinatorial framework for identifying proteolytic E3-substrate relationships
at scale. Deep-learning-base...
www.cell.com
February 9, 2025 at 6:53 PM
Combinatorial mapping of E3 ubiquitin ligases to their target substrates
www.cell.com/molecular-ce...
www.cell.com/molecular-ce...
Reposted by Xi Fu

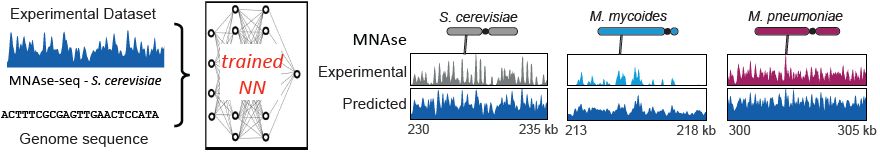
Deep learning models (@chromozz.bsky.social) trained only on yeast chromosomes predict nucleosome positioning, RNA Poll II and cohesin tracks along foreign DNA, based on the sequence alone. This implies that the behavior of any DNA in a host cell follows deterministic sequence-based rules.
February 7, 2025 at 10:22 AM
Deep learning models (@chromozz.bsky.social) trained only on yeast chromosomes predict nucleosome positioning, RNA Poll II and cohesin tracks along foreign DNA, based on the sequence alone. This implies that the behavior of any DNA in a host cell follows deterministic sequence-based rules.
Reposted by Xi Fu

[SAVE THE DATE] MLCB 2025 is happening Sept 10-11 at the NY Genome Center in NYC!
Attend the premier conference at the intersection of ML & Bio, share your research and make lasting connections!
Submission deadline: June 1
More details: mlcb.github.io
Help spread the word—please RT! #MLCB2025
Attend the premier conference at the intersection of ML & Bio, share your research and make lasting connections!
Submission deadline: June 1
More details: mlcb.github.io
Help spread the word—please RT! #MLCB2025
February 5, 2025 at 2:50 AM
[SAVE THE DATE] MLCB 2025 is happening Sept 10-11 at the NY Genome Center in NYC!
Attend the premier conference at the intersection of ML & Bio, share your research and make lasting connections!
Submission deadline: June 1
More details: mlcb.github.io
Help spread the word—please RT! #MLCB2025
Attend the premier conference at the intersection of ML & Bio, share your research and make lasting connections!
Submission deadline: June 1
More details: mlcb.github.io
Help spread the word—please RT! #MLCB2025
Reposted by Xi Fu

Lars Steinmetz and @seczmarta.bsky.social put together a wonderful perspective on these two studies. www.science.org/doi/10.1126/...
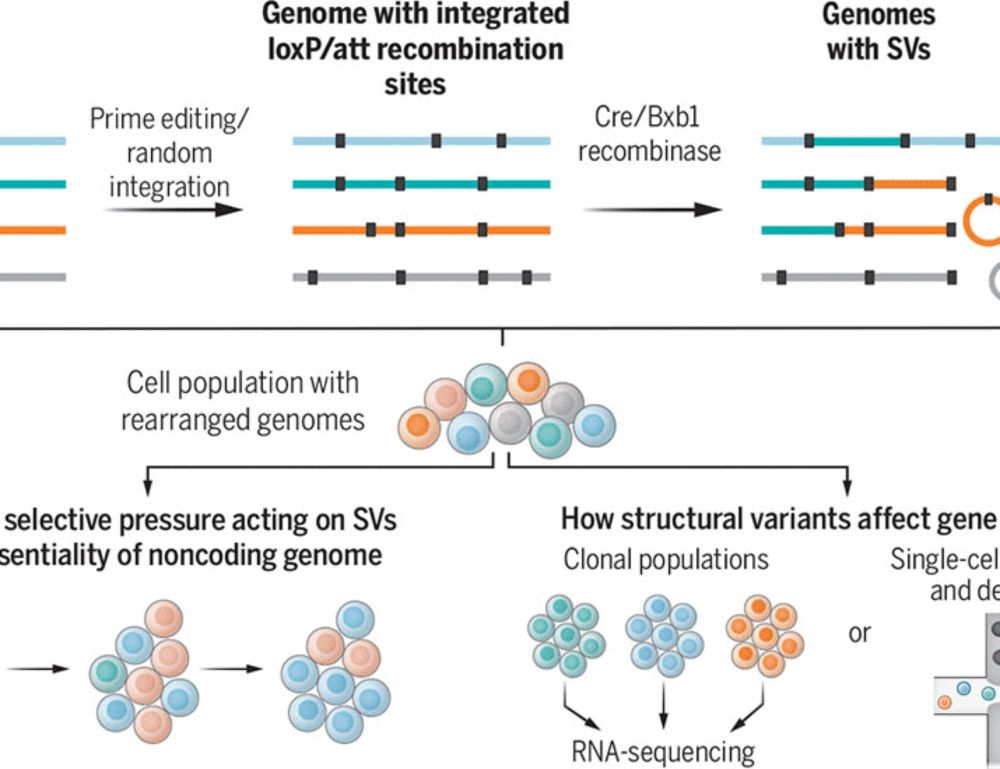
Genome recombination on demand
Large genome rearrangements in mammalian cells can be generated at scale
www.science.org
January 31, 2025 at 1:49 PM
Lars Steinmetz and @seczmarta.bsky.social put together a wonderful perspective on these two studies. www.science.org/doi/10.1126/...
Reposted by Xi Fu

The "kitchen sink" of omics to solve the basis for an undiagnosed disease: long read genome , transcriptome, methytome, epigenome, all synchronized (a first)
www.nature.com/articles/s41...
www.nature.com/articles/s41...
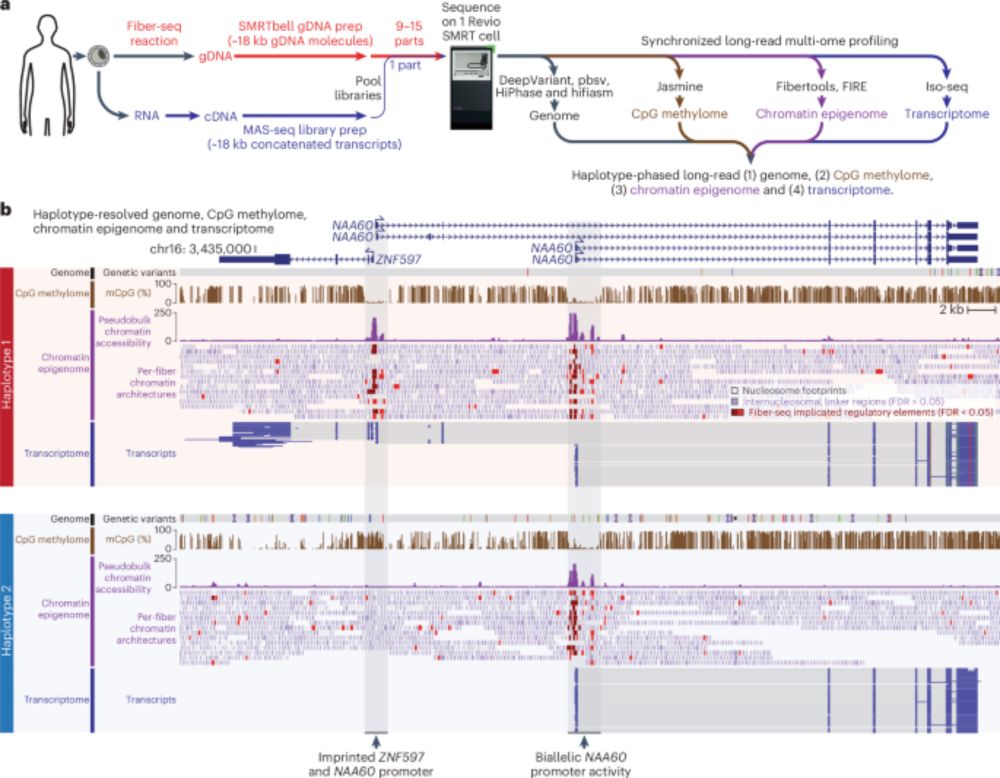
Synchronized long-read genome, methylome, epigenome and transcriptome profiling resolve a Mendelian condition - Nature Genetics
Simultaneous profiling of the genome, methylome, epigenome and transcriptome using single-molecule chromatin fiber sequencing and multiplexed arrays isoform sequencing identifies the genetic and molec...
www.nature.com
January 29, 2025 at 2:45 PM
The "kitchen sink" of omics to solve the basis for an undiagnosed disease: long read genome , transcriptome, methytome, epigenome, all synchronized (a first)
www.nature.com/articles/s41...
www.nature.com/articles/s41...
Reposted by Xi Fu

Super excited to share our new study from the @jbuenrostro.bsky.social Lab in @nature.com! We developed a computational method for tracking transcription factor and nucleosome binding using single-cell ATAC-seq and deep learning.
Paper: www.nature.com/articles/s41...
Paper: www.nature.com/articles/s41...
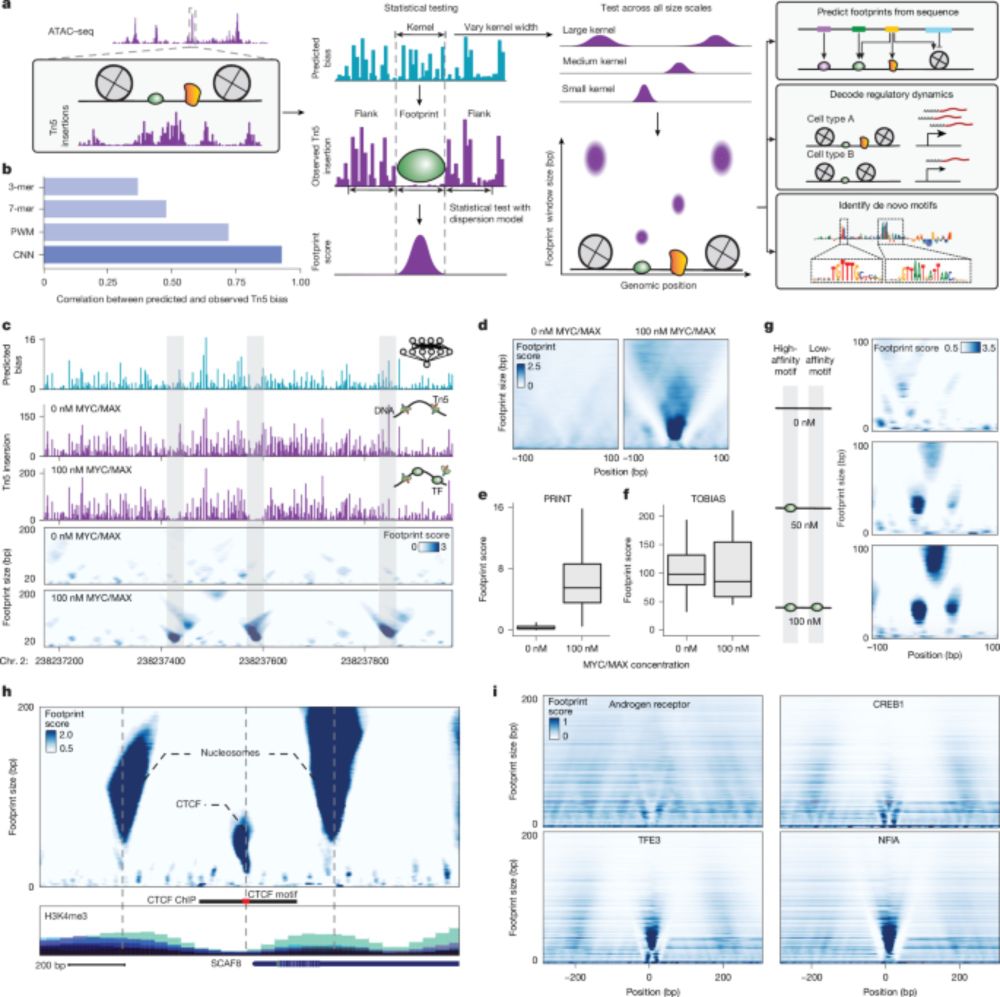
Multiscale footprints reveal the organization of cis-regulatory elements - Nature
We developed PRINT, a computational method that identifies footprints of DNA–protein interactions from bulk and single-cell chromatin accessibility data across multiple scales of protein size.
www.nature.com
January 23, 2025 at 2:11 AM
Super excited to share our new study from the @jbuenrostro.bsky.social Lab in @nature.com! We developed a computational method for tracking transcription factor and nucleosome binding using single-cell ATAC-seq and deep learning.
Paper: www.nature.com/articles/s41...
Paper: www.nature.com/articles/s41...
The most senior cell typing expert should and always have been the evolution

I've heard that in clinical pathology, ground truth is whatever the most senior pathologist says it is. Kinda reminds me of cell typing.
January 16, 2025 at 8:11 PM
The most senior cell typing expert should and always have been the evolution
Reposted by Xi Fu

@anusri.bsky.social first author & developer of ChromBPNet is looking for opportunities in industry in ML for bio/genomics. She is an excellent rigorous scientist (as u can see from the paper). Very strongly recommend her. Plz reach out to her if u have openings. Plz forward.
Our original biorxiv submission of the ChromBPNet preprint had issues with supp. methods & file links not working (even though we they were uploaded). This updated version has fixed those issues. Everything shud be available now. Thanks for your patience.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
January 13, 2025 at 6:23 PM
@anusri.bsky.social first author & developer of ChromBPNet is looking for opportunities in industry in ML for bio/genomics. She is an excellent rigorous scientist (as u can see from the paper). Very strongly recommend her. Plz reach out to her if u have openings. Plz forward.
Reposted by Xi Fu

The human genome encodes more than 20,000 proteins. Missense variants in nearly 5,000 of these proteins cause Mendelian diseases. Most variants compatible with life are likely present in someone currently alive. The study marks an important step in understanding the functional consequences.

Site-saturation mutagenesis of 500 human protein domains - Nature
Large-scale experimental analysis of Human Domainome 1, a library containing more than 500,000 missense mutation variants across more than 500 human protein domains, reveals that 60% of pathogenic mis...
www.nature.com
January 8, 2025 at 4:09 PM
The human genome encodes more than 20,000 proteins. Missense variants in nearly 5,000 of these proteins cause Mendelian diseases. Most variants compatible with life are likely present in someone currently alive. The study marks an important step in understanding the functional consequences.
GET is finally published!
- Paper: t.ly/iQct_ (new validations, dry and wet)
- Model: t.ly/4jnUI (new tutorial on PBMC 10x Multiome data, and yes you can even fine-tune it on a Macbook)
- Analysis package: t.ly/OqLAL
- Demo: t.ly/rbFQB
- Docker: t.ly/86n_i
- Paper: t.ly/iQct_ (new validations, dry and wet)
- Model: t.ly/4jnUI (new tutorial on PBMC 10x Multiome data, and yes you can even fine-tune it on a Macbook)
- Analysis package: t.ly/OqLAL
- Demo: t.ly/rbFQB
- Docker: t.ly/86n_i
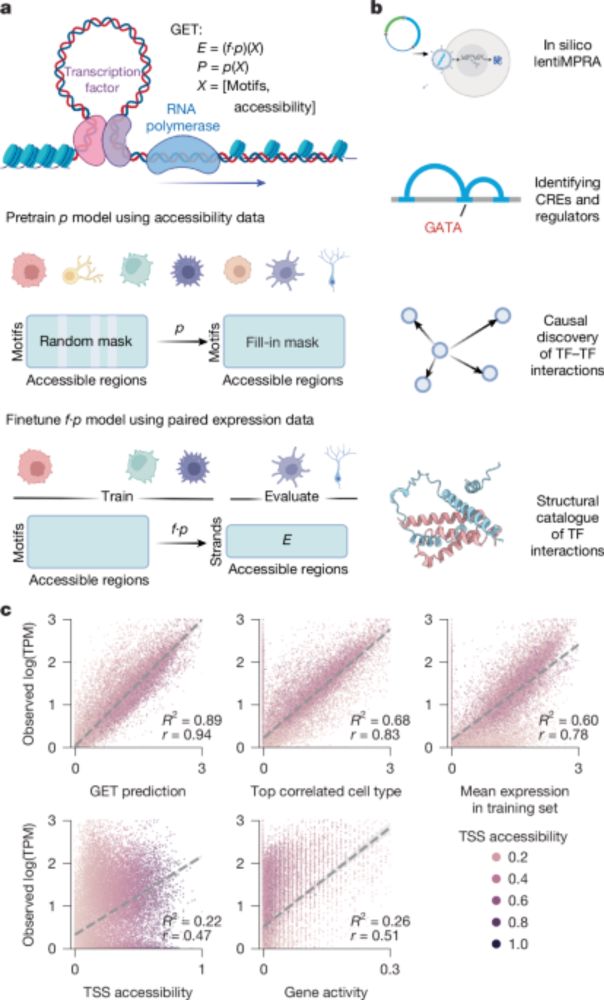
A foundation model of transcription across human cell types - Nature
A foundation model learns transcriptional regulatory syntax from chromatin accessibility and sequence data across a range of cell types to predict gene expression and transcription factor interactions...
t.ly
January 8, 2025 at 4:16 PM
GET is finally published!
- Paper: t.ly/iQct_ (new validations, dry and wet)
- Model: t.ly/4jnUI (new tutorial on PBMC 10x Multiome data, and yes you can even fine-tune it on a Macbook)
- Analysis package: t.ly/OqLAL
- Demo: t.ly/rbFQB
- Docker: t.ly/86n_i
- Paper: t.ly/iQct_ (new validations, dry and wet)
- Model: t.ly/4jnUI (new tutorial on PBMC 10x Multiome data, and yes you can even fine-tune it on a Macbook)
- Analysis package: t.ly/OqLAL
- Demo: t.ly/rbFQB
- Docker: t.ly/86n_i
Reposted by Xi Fu
Our ChromBPNet preprint out!
www.biorxiv.org/content/10.1...
Huge congrats to Anusri! This was quite a slog (for both of us) but we r very proud of this one! It is a long read but worth it IMHO. Methods r in the supp. materials. Bluetorial coming soon below 1/
www.biorxiv.org/content/10.1...
Huge congrats to Anusri! This was quite a slog (for both of us) but we r very proud of this one! It is a long read but worth it IMHO. Methods r in the supp. materials. Bluetorial coming soon below 1/
December 25, 2024 at 11:48 PM
Our ChromBPNet preprint out!
www.biorxiv.org/content/10.1...
Huge congrats to Anusri! This was quite a slog (for both of us) but we r very proud of this one! It is a long read but worth it IMHO. Methods r in the supp. materials. Bluetorial coming soon below 1/
www.biorxiv.org/content/10.1...
Huge congrats to Anusri! This was quite a slog (for both of us) but we r very proud of this one! It is a long read but worth it IMHO. Methods r in the supp. materials. Bluetorial coming soon below 1/
Reposted by Xi Fu

What do GWAS and rare variant burden tests discover, and why?
Do these studies find the most IMPORTANT genes? If not, how DO they rank genes?
Here we present a surprising result: these studies actually test for SPECIFICITY! A 🧵on what this means... (🧪🧬)
www.biorxiv.org/content/10.1...
Do these studies find the most IMPORTANT genes? If not, how DO they rank genes?
Here we present a surprising result: these studies actually test for SPECIFICITY! A 🧵on what this means... (🧪🧬)
www.biorxiv.org/content/10.1...

Specificity, length, and luck: How genes are prioritized by rare and common variant association studies
Standard genome-wide association studies (GWAS) and rare variant burden tests are essential tools for identifying trait-relevant genes. Although these methods are conceptually similar, we show by anal...
www.biorxiv.org
December 17, 2024 at 7:05 AM
What do GWAS and rare variant burden tests discover, and why?
Do these studies find the most IMPORTANT genes? If not, how DO they rank genes?
Here we present a surprising result: these studies actually test for SPECIFICITY! A 🧵on what this means... (🧪🧬)
www.biorxiv.org/content/10.1...
Do these studies find the most IMPORTANT genes? If not, how DO they rank genes?
Here we present a surprising result: these studies actually test for SPECIFICITY! A 🧵on what this means... (🧪🧬)
www.biorxiv.org/content/10.1...