



Sergio Mares
@sergiomares.bsky.social
/sˈeɾ.xjo/ | Computational Biology PhD Candidate | @UCBerkeley-@UCSFCancer | genomics, cancer and ml | 🇲🇽
Reposted by Sergio Mares

If you’re a federal govt employee targeted for leave or termination on the basis of your DEI work, please file an EEO (Equal Employment Opportunity) complaint & retain legal counsel.
I’m a former NIH PI. Happy to refer you to a law firm that has successfully sued the govt many times!
Pls repost 🙏❤️
I’m a former NIH PI. Happy to refer you to a law firm that has successfully sued the govt many times!
Pls repost 🙏❤️
January 24, 2025 at 10:02 AM
If you’re a federal govt employee targeted for leave or termination on the basis of your DEI work, please file an EEO (Equal Employment Opportunity) complaint & retain legal counsel.
I’m a former NIH PI. Happy to refer you to a law firm that has successfully sued the govt many times!
Pls repost 🙏❤️
I’m a former NIH PI. Happy to refer you to a law firm that has successfully sued the govt many times!
Pls repost 🙏❤️
Walked out of a 2 hr meeting with my PI feeling both refreshed and a bit confused about the future
January 25, 2025 at 2:39 AM
Walked out of a 2 hr meeting with my PI feeling both refreshed and a bit confused about the future
Reposted by Sergio Mares

And there it goes. The PAR for Research Supplements to Promote Diversity in Health-Related Research has been updated to reflect a new "Expiration Date: New Date January 24, 2025 (Original Expiration Date: May 8, 2026)"
grants.nih.gov/grants/guide...
grants.nih.gov/grants/guide...

January 24, 2025 at 10:43 PM
And there it goes. The PAR for Research Supplements to Promote Diversity in Health-Related Research has been updated to reflect a new "Expiration Date: New Date January 24, 2025 (Original Expiration Date: May 8, 2026)"
grants.nih.gov/grants/guide...
grants.nih.gov/grants/guide...
Reposted by Sergio Mares

Congrats to the @jbuenrostro.bsky.social lab on this recent paper.
www.nature.com/articles/s41...
Unfortunately, it makes several misleading claims that we disagree with. We will address these technical & other issues with this work over the next few months.
www.nature.com/articles/s41...
Unfortunately, it makes several misleading claims that we disagree with. We will address these technical & other issues with this work over the next few months.

Multiscale footprints reveal the organization of cis-regulatory elements - Nature
We developed PRINT, a computational method that identifies footprints of DNA–protein interactions from bulk and single-cell chromatin accessibility data across multiple scales of protein size.
www.nature.com
January 23, 2025 at 7:18 PM
Congrats to the @jbuenrostro.bsky.social lab on this recent paper.
www.nature.com/articles/s41...
Unfortunately, it makes several misleading claims that we disagree with. We will address these technical & other issues with this work over the next few months.
www.nature.com/articles/s41...
Unfortunately, it makes several misleading claims that we disagree with. We will address these technical & other issues with this work over the next few months.
Reposted by Sergio Mares

Check out our new & improved V2 manuscript for RiboNN, with new analyses to further sharpen the results!! www.biorxiv.org/content/10.1...
January 20, 2025 at 1:57 PM
Check out our new & improved V2 manuscript for RiboNN, with new analyses to further sharpen the results!! www.biorxiv.org/content/10.1...
Reposted by Sergio Mares

All NIH study sections canceled indefinitely. This will halt science and devastate research budgets in universities.
January 22, 2025 at 8:46 PM
All NIH study sections canceled indefinitely. This will halt science and devastate research budgets in universities.

Reposted by Sergio Mares

Super excited to share our review on genomic deep learning models for non-coding variant effect prediction, with Ayesha Bajwa and Nilah Ioannidis. We’d like this review to be a useful resource, and welcome any feedback, comments, or questions! 1/4
arxiv.org/abs/2411.11158
arxiv.org/abs/2411.11158

Leveraging genomic deep learning models for non-coding variant effect prediction
The majority of genetic variants identified in genome-wide association studies of complex traits are non-coding, and characterizing their function remains an important challenge in human genetics. Gen...
arxiv.org
November 20, 2024 at 1:31 AM
Super excited to share our review on genomic deep learning models for non-coding variant effect prediction, with Ayesha Bajwa and Nilah Ioannidis. We’d like this review to be a useful resource, and welcome any feedback, comments, or questions! 1/4
arxiv.org/abs/2411.11158
arxiv.org/abs/2411.11158
Reposted by Sergio Mares

Who's going to be at NeurIPS this year? We'll be there all week and presenting some of our work on assessing protein-peptide interactions at the MLSB workshop. Come say hi if you're around!
Paper: www.biorxiv.org/content/10.1...
Paper: www.biorxiv.org/content/10.1...

November 19, 2024 at 9:23 AM
Who's going to be at NeurIPS this year? We'll be there all week and presenting some of our work on assessing protein-peptide interactions at the MLSB workshop. Come say hi if you're around!
Paper: www.biorxiv.org/content/10.1...
Paper: www.biorxiv.org/content/10.1...
Reposted by Sergio Mares

Many reporter assays assume that the determinants of expression are 1) DNA sequence and 2) trans-acting soluble factors. It's ambitious but natural to want to model dose response to regulators as a function of DNA sequence, and I'm excited to see this.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...

Transfer learning reveals sequence determinants of the quantitative response to transcription factor dosage
Deep learning approaches have made significant advances in predicting cell type-specific chromatin patterns from the identity and arrangement of transcription factor (TF) binding motifs. However, most...
www.biorxiv.org
November 18, 2024 at 5:00 PM
Many reporter assays assume that the determinants of expression are 1) DNA sequence and 2) trans-acting soluble factors. It's ambitious but natural to want to model dose response to regulators as a function of DNA sequence, and I'm excited to see this.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
Reposted by Sergio Mares

Large protein language models can learn complex epistatic interactions, but how much does that help with predicting variant effects? In this NeurIPS article, we show that classical independent-sites phylogenetic models can outperform pLMs on this task.
1/7
openreview.net/forum?id=H7m...
1/7
openreview.net/forum?id=H7m...

Ultrafast classical phylogenetic method beats large protein...
Amino acid substitution rate matrices are fundamental to statistical phylogenetics and evolutionary biology. Estimating them typically requires reconstructed trees for massive amounts of aligned...
openreview.net
November 16, 2024 at 8:42 PM
Large protein language models can learn complex epistatic interactions, but how much does that help with predicting variant effects? In this NeurIPS article, we show that classical independent-sites phylogenetic models can outperform pLMs on this task.
1/7
openreview.net/forum?id=H7m...
1/7
openreview.net/forum?id=H7m...
Reposted by Sergio Mares

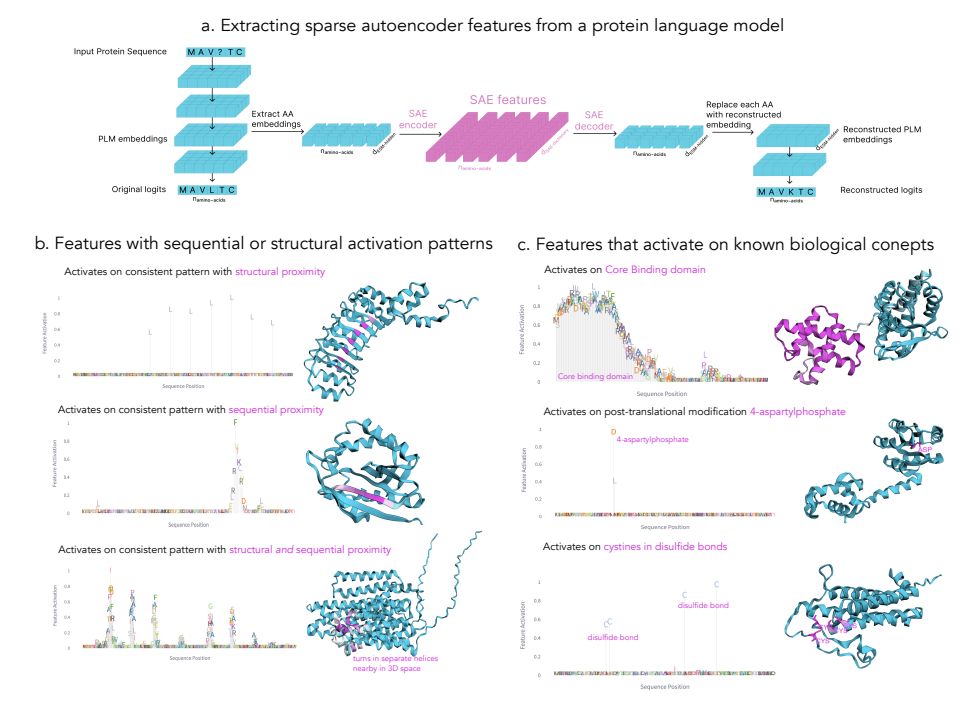
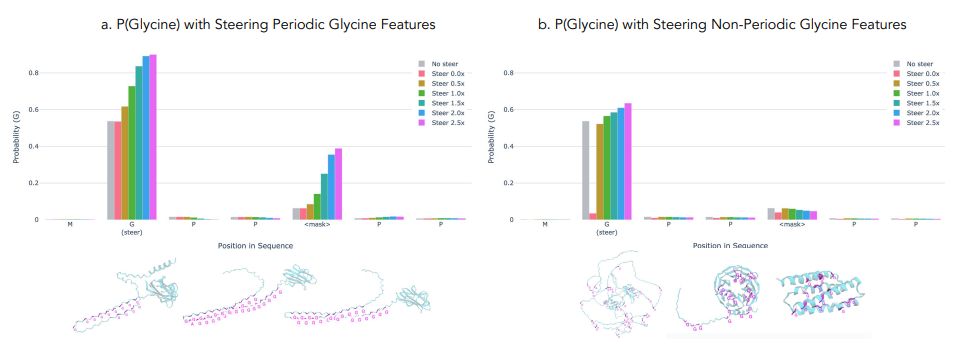
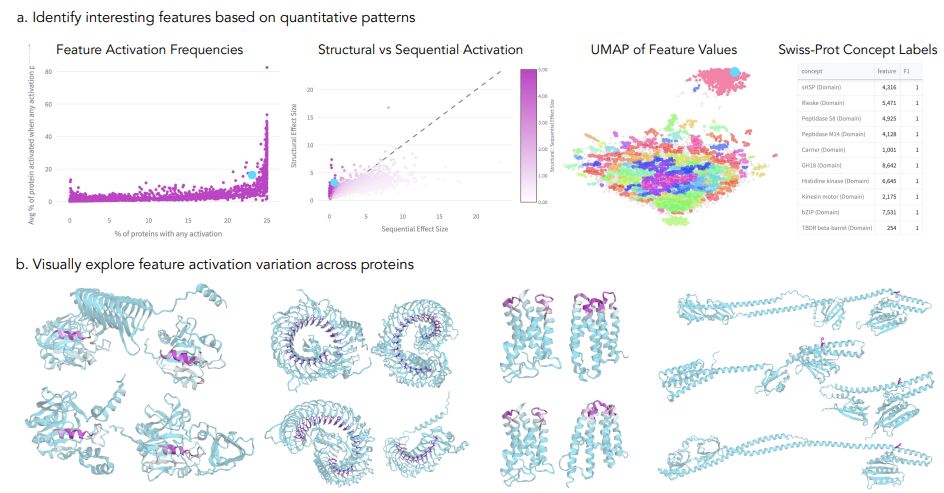
November 18, 2024 at 10:20 PM
Reposted by Sergio Mares

Thrilled to announce Boltz-1, the first open-source and commercially available model to achieve AlphaFold3-level accuracy on biomolecular structure prediction! An exciting collaboration with Jeremy, Saro, and an amazing team at MIT and Genesis Therapeutics. A thread!
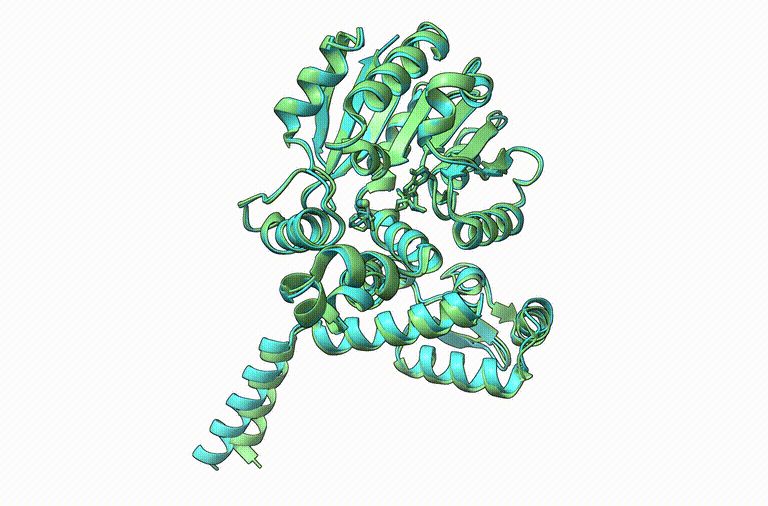
November 17, 2024 at 4:20 PM
Thrilled to announce Boltz-1, the first open-source and commercially available model to achieve AlphaFold3-level accuracy on biomolecular structure prediction! An exciting collaboration with Jeremy, Saro, and an amazing team at MIT and Genesis Therapeutics. A thread!
Reposted by Sergio Mares
Boltz-1: Open source (MIT license) protein + PPI + PLI structure prediction method performs on par with AlphaFold3 by Gabriel Corso, Barzilay et al. & Genesis Therapeutics
Blog-post jclinic.mit.edu/boltz-1/
Technical report: t.co/px0spnTA0S
Model and code: github.com/jwohlwend/boltz
Blog-post jclinic.mit.edu/boltz-1/
Technical report: t.co/px0spnTA0S
Model and code: github.com/jwohlwend/boltz
Introducing Boltz-1: Democratizing Biomolecular Interaction Modeling – MIT Jameel Clinic
jclinic.mit.edu
November 18, 2024 at 1:37 AM
Boltz-1: Open source (MIT license) protein + PPI + PLI structure prediction method performs on par with AlphaFold3 by Gabriel Corso, Barzilay et al. & Genesis Therapeutics
Blog-post jclinic.mit.edu/boltz-1/
Technical report: t.co/px0spnTA0S
Model and code: github.com/jwohlwend/boltz
Blog-post jclinic.mit.edu/boltz-1/
Technical report: t.co/px0spnTA0S
Model and code: github.com/jwohlwend/boltz
Reposted by Sergio Mares

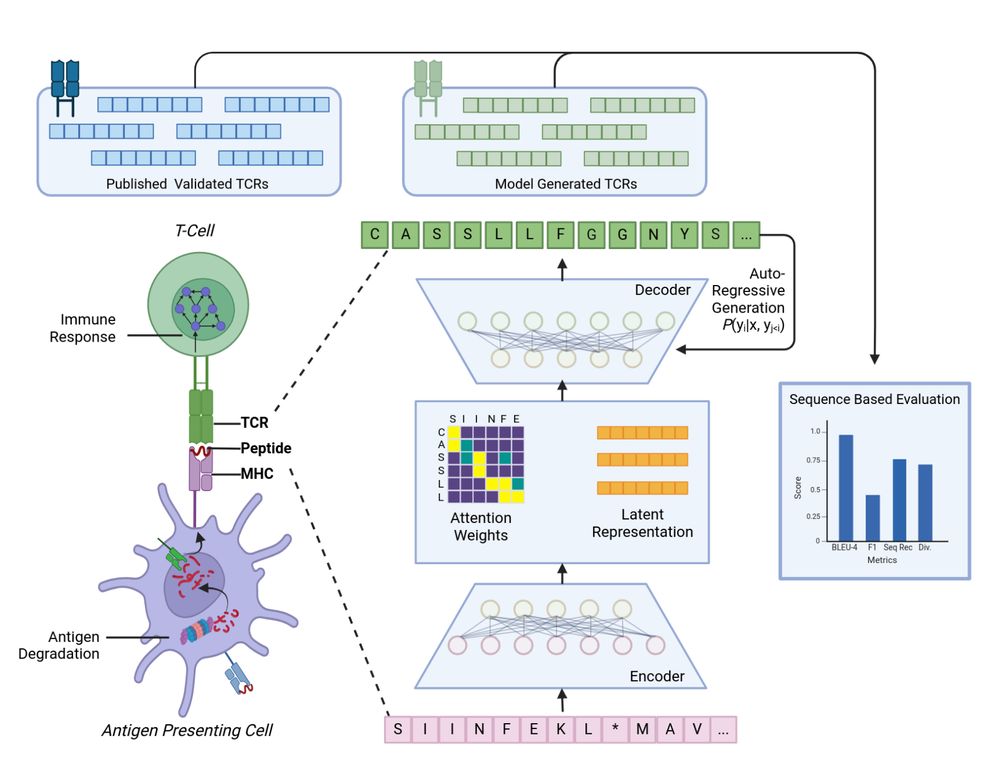
New de-novo TCR design preprint from @dkarthikey1.bsky.social just dropped:
TCR-TRANSLATE: Conditional Generation of Real Antigen Specific T-cell Receptor Sequences
www.biorxiv.org/content/10.1...
TCR-TRANSLATE: Conditional Generation of Real Antigen Specific T-cell Receptor Sequences
www.biorxiv.org/content/10.1...
November 13, 2024 at 3:29 AM
New de-novo TCR design preprint from @dkarthikey1.bsky.social just dropped:
TCR-TRANSLATE: Conditional Generation of Real Antigen Specific T-cell Receptor Sequences
www.biorxiv.org/content/10.1...
TCR-TRANSLATE: Conditional Generation of Real Antigen Specific T-cell Receptor Sequences
www.biorxiv.org/content/10.1...