



Kaitlin Samocha
@ksamocha.bsky.social
Assistant Investigator @ MGH / Broad / HMS. Focus on human genomics and modeling rare variation. She/her
Reposted by Kaitlin Samocha

New paper on everyone’s favourite topic, QC!
We show why you should do genotype-level QC on your WGS data
www.biorxiv.org/content/10.1...
Very real quotes about this paper -
“The most exciting, mind-blowing paper of the year!”
“On a par with Fisher 1918”
“I read it every night. Just so beautiful”
We show why you should do genotype-level QC on your WGS data
www.biorxiv.org/content/10.1...
Very real quotes about this paper -
“The most exciting, mind-blowing paper of the year!”
“On a par with Fisher 1918”
“I read it every night. Just so beautiful”

Genotype-level quality control substantially reduces error rates in population-scale whole-genome sequencing
Population-scale whole-genome sequencing data will contain many individual-level genotype errors, even after allele-level quality control (QC). We establish the need for genotype-level QC using UK Bio...
www.biorxiv.org
November 8, 2025 at 9:31 AM
New paper on everyone’s favourite topic, QC!
We show why you should do genotype-level QC on your WGS data
www.biorxiv.org/content/10.1...
Very real quotes about this paper -
“The most exciting, mind-blowing paper of the year!”
“On a par with Fisher 1918”
“I read it every night. Just so beautiful”
We show why you should do genotype-level QC on your WGS data
www.biorxiv.org/content/10.1...
Very real quotes about this paper -
“The most exciting, mind-blowing paper of the year!”
“On a par with Fisher 1918”
“I read it every night. Just so beautiful”
Reposted by Kaitlin Samocha

New study of 800K+ genomes from gnomAD reveals most “pathogenic” variants in healthy people aren’t truly disease-tolerant. They are explained by annotation errors, mosaicism, or compensatory variants. 🧬
A big step for precision medicine!
www.nature.com/articles/s41...
A big step for precision medicine!
www.nature.com/articles/s41...

Exploring penetrance of clinically relevant variants in over 800,000 humans from the Genome Aggregation Database - Nature Communications
Here the authors provide an explanation for 95% of examined predicted loss of function variants found in disease-associated haploinsufficient genes in the Genome Aggregation Database (gnomAD),…
www.nature.com
November 4, 2025 at 3:06 PM
New study of 800K+ genomes from gnomAD reveals most “pathogenic” variants in healthy people aren’t truly disease-tolerant. They are explained by annotation errors, mosaicism, or compensatory variants. 🧬
A big step for precision medicine!
www.nature.com/articles/s41...
A big step for precision medicine!
www.nature.com/articles/s41...
Reposted by Kaitlin Samocha

📃 We’re excited to share our latest work, now published in Nature Communications — a major update to the Genome Aggregation Database (gnomAD) that improves allele frequency resolution for two gnomAD-defined genetic ancestry groups using local ancestry inference (LAI).
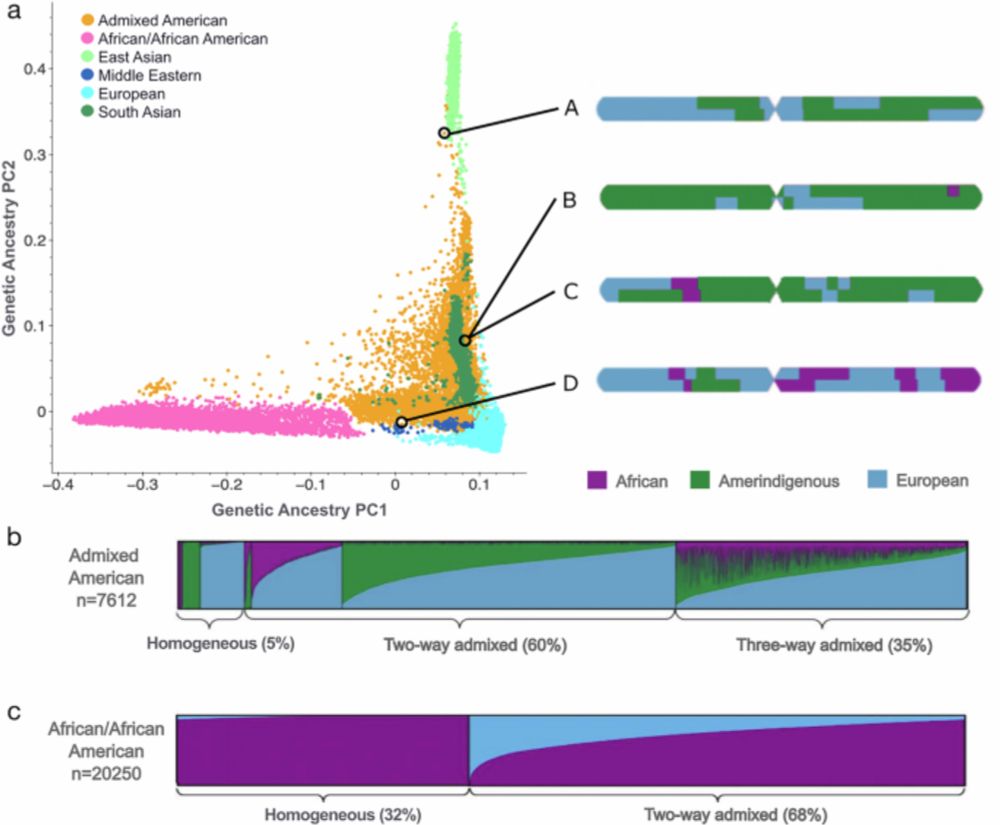
Improved allele frequencies in gnomAD through local ancestry inference - Nature Communications
This study incorporates local ancestry into the Genome Aggregation Database (gnomAD) to improve allele frequency estimates for admixed populations, enhancing variant interpretation and enabling more accurate and equitable genomic research and clinical care.
www.nature.com
October 6, 2025 at 6:31 PM
📃 We’re excited to share our latest work, now published in Nature Communications — a major update to the Genome Aggregation Database (gnomAD) that improves allele frequency resolution for two gnomAD-defined genetic ancestry groups using local ancestry inference (LAI).
Reposted by Kaitlin Samocha

Now published! Our paper on:
(1) Accurate sequencing of sperm at scale
(2) Positive selection of spermatogenesis driver mutations across the exome
(3) Offspring disease risks from male reproductive aging
[1/n]
www.nature.com/articles/s41...
(1) Accurate sequencing of sperm at scale
(2) Positive selection of spermatogenesis driver mutations across the exome
(3) Offspring disease risks from male reproductive aging
[1/n]
www.nature.com/articles/s41...

Sperm sequencing reveals extensive positive selection in the male germline - Nature
A combination of whole-genome NanoSeq with deep whole-exome and targeted NanoSeq is used to accurately characterize mutation rates and genes under positive selection in sperm cells.
www.nature.com
October 8, 2025 at 3:51 PM
Now published! Our paper on:
(1) Accurate sequencing of sperm at scale
(2) Positive selection of spermatogenesis driver mutations across the exome
(3) Offspring disease risks from male reproductive aging
[1/n]
www.nature.com/articles/s41...
(1) Accurate sequencing of sperm at scale
(2) Positive selection of spermatogenesis driver mutations across the exome
(3) Offspring disease risks from male reproductive aging
[1/n]
www.nature.com/articles/s41...
Reposted by Kaitlin Samocha

📣 We are recruiting! Please share!!
Are you a bioinformatician / computational scientist who wants to apply your skills to understanding regulatory biology and improving rare disease diagnosis and treatment? 🧠 💻 🧬 🩺
We have two roles available 👇
🧵 1/4
Are you a bioinformatician / computational scientist who wants to apply your skills to understanding regulatory biology and improving rare disease diagnosis and treatment? 🧠 💻 🧬 🩺
We have two roles available 👇
🧵 1/4

July 31, 2025 at 4:12 PM
📣 We are recruiting! Please share!!
Are you a bioinformatician / computational scientist who wants to apply your skills to understanding regulatory biology and improving rare disease diagnosis and treatment? 🧠 💻 🧬 🩺
We have two roles available 👇
🧵 1/4
Are you a bioinformatician / computational scientist who wants to apply your skills to understanding regulatory biology and improving rare disease diagnosis and treatment? 🧠 💻 🧬 🩺
We have two roles available 👇
🧵 1/4
Reposted by Kaitlin Samocha
Isn't genetics cool???
Within only 145 nucleotides(!) of a non-coding RNA (RNU4-2) - different variants in distinct regions / structures cause three distinct disorders!!! (all discovered within the last 18 months)
🤯🤓🧬❤️
Within only 145 nucleotides(!) of a non-coding RNA (RNU4-2) - different variants in distinct regions / structures cause three distinct disorders!!! (all discovered within the last 18 months)
🤯🤓🧬❤️

August 18, 2025 at 2:03 PM
Isn't genetics cool???
Within only 145 nucleotides(!) of a non-coding RNA (RNU4-2) - different variants in distinct regions / structures cause three distinct disorders!!! (all discovered within the last 18 months)
🤯🤓🧬❤️
Within only 145 nucleotides(!) of a non-coding RNA (RNU4-2) - different variants in distinct regions / structures cause three distinct disorders!!! (all discovered within the last 18 months)
🤯🤓🧬❤️
Reposted by Kaitlin Samocha

🗣️ Quote of #ESHG2025 (so far)
"Who licks bone !?!" 🦴
- Johannes Krause
Anyone have that on your bingo card?
Well apparently archeologists do, to distinguish bone from stones and it causes problems in DNA sequencing. 🤔
"Who licks bone !?!" 🦴
- Johannes Krause
Anyone have that on your bingo card?
Well apparently archeologists do, to distinguish bone from stones and it causes problems in DNA sequencing. 🤔

May 24, 2025 at 1:46 PM
🗣️ Quote of #ESHG2025 (so far)
"Who licks bone !?!" 🦴
- Johannes Krause
Anyone have that on your bingo card?
Well apparently archeologists do, to distinguish bone from stones and it causes problems in DNA sequencing. 🤔
"Who licks bone !?!" 🦴
- Johannes Krause
Anyone have that on your bingo card?
Well apparently archeologists do, to distinguish bone from stones and it causes problems in DNA sequencing. 🤔
We are just wrapping up day 1 at #ESGH2025 in beautiful Milan. For those who want some extra fun while listening to the great science, you can play bingo.👇
I know multiple of these have already occurred.
I know multiple of these have already occurred.
Getting ready for #eshg2025 …and our postdoc @lydiasagath.bsky.social made a nice BINGO card again.
Paying a lot of attention to the entire event will pay off!
Paying a lot of attention to the entire event will pay off!

May 24, 2025 at 5:14 PM
We are just wrapping up day 1 at #ESGH2025 in beautiful Milan. For those who want some extra fun while listening to the great science, you can play bingo.👇
I know multiple of these have already occurred.
I know multiple of these have already occurred.
Reposted by Kaitlin Samocha

Buongiorno Milano! Ready for a great day 1 of #eshg2025?
Packed program of excellent science 8.30am-8.00pm - plus networking event till 9.30pm to meet many friends, colleagues and collaborators! …andiamo @eshg.bsky.social @eshgyoung.bsky.social
Packed program of excellent science 8.30am-8.00pm - plus networking event till 9.30pm to meet many friends, colleagues and collaborators! …andiamo @eshg.bsky.social @eshgyoung.bsky.social
May 24, 2025 at 4:45 AM
Buongiorno Milano! Ready for a great day 1 of #eshg2025?
Packed program of excellent science 8.30am-8.00pm - plus networking event till 9.30pm to meet many friends, colleagues and collaborators! …andiamo @eshg.bsky.social @eshgyoung.bsky.social
Packed program of excellent science 8.30am-8.00pm - plus networking event till 9.30pm to meet many friends, colleagues and collaborators! …andiamo @eshg.bsky.social @eshgyoung.bsky.social
Reposted by Kaitlin Samocha

🤗 Hugely excited to share our work on automating iterative reanalysis in #raredisease, preprint out: www.medrxiv.org/content/10.1... 🤖🧬
github.com/populationge...
A superb collaboration with @dgmacarthur.bsky.social @cassimons.bsky.social @heidirehm.bsky.social @ksamocha.bsky.social and many more!
github.com/populationge...
A superb collaboration with @dgmacarthur.bsky.social @cassimons.bsky.social @heidirehm.bsky.social @ksamocha.bsky.social and many more!

Scalable automated reanalysis of genomic data in research and clinical rare disease cohorts
Reanalysis of genomic data in rare disease is highly effective in increasing diagnostic yields but remains limited by manual approaches. Automation and optimization for high specificity will be necess...
www.medrxiv.org
May 23, 2025 at 6:04 AM
🤗 Hugely excited to share our work on automating iterative reanalysis in #raredisease, preprint out: www.medrxiv.org/content/10.1... 🤖🧬
github.com/populationge...
A superb collaboration with @dgmacarthur.bsky.social @cassimons.bsky.social @heidirehm.bsky.social @ksamocha.bsky.social and many more!
github.com/populationge...
A superb collaboration with @dgmacarthur.bsky.social @cassimons.bsky.social @heidirehm.bsky.social @ksamocha.bsky.social and many more!
Reposted by Kaitlin Samocha

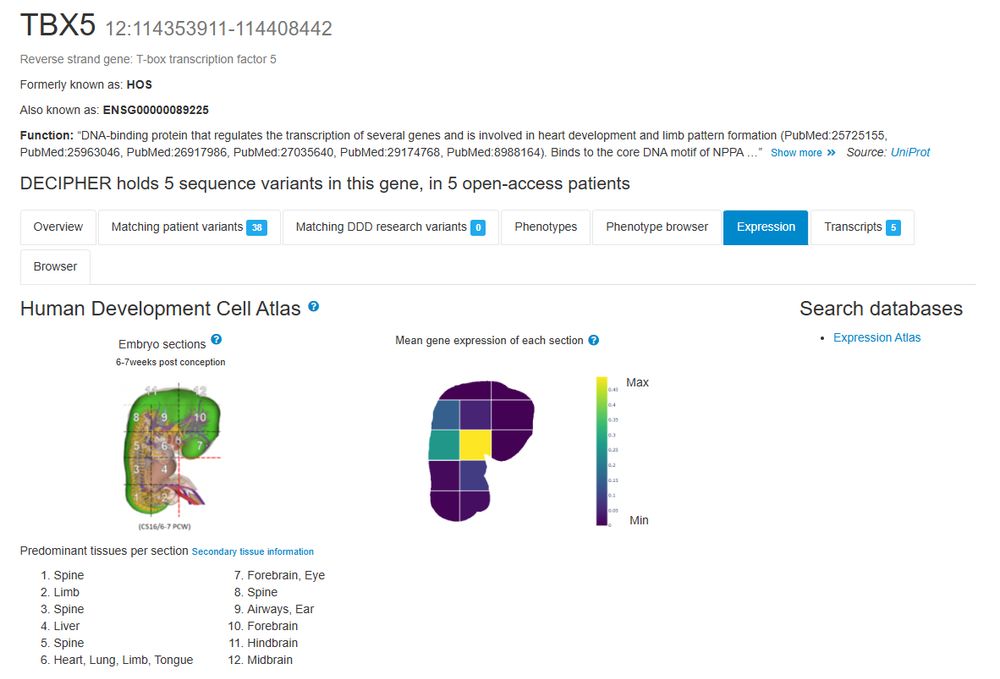
Human Developmental Cell Atlas (HDCA) expression data is now displayed. Expression is displayed in 12 sections of a 6-7 post-conception week human embryo, alongside a sagittal view which displays the region of the embryo represented by each section @mhaniffa.bsky.social
May 7, 2025 at 1:29 PM
Human Developmental Cell Atlas (HDCA) expression data is now displayed. Expression is displayed in 12 sections of a 6-7 post-conception week human embryo, alongside a sagittal view which displays the region of the embryo represented by each section @mhaniffa.bsky.social
Reposted by Kaitlin Samocha
A few weeks ago, I had an incredibly emotional call with James Coney, a writer for the Sunday Times whose son Charlie was in the @genomicsengland.bsky.social 100k project and was recently diagnosed with ReNU syndrome. This beautiful article tells their story ❤️ www.thetimes.com/article/0bcc...

My son Charlie — and the breakthrough that changed our lives
James Coney and his wife, Sarah, struggled not knowing why their 12-year-old was born with a severe learning disability. In their darkest moments, they blamed themselves. Then, out of the blue, came a...
www.thetimes.com
March 2, 2025 at 12:06 PM
A few weeks ago, I had an incredibly emotional call with James Coney, a writer for the Sunday Times whose son Charlie was in the @genomicsengland.bsky.social 100k project and was recently diagnosed with ReNU syndrome. This beautiful article tells their story ❤️ www.thetimes.com/article/0bcc...
Reposted by Kaitlin Samocha

Join leading experts working in #RareDisease research at our #GRD25 conference.
📅 Dates: 9-11 April 2025
💭 Share insights in person
Explore the latest #genomics advances accelerating improvements in clinical care for rare disorders, globally.
⏰Secure your place by 11 March: bit.ly/3BpAe44
📅 Dates: 9-11 April 2025
💭 Share insights in person
Explore the latest #genomics advances accelerating improvements in clinical care for rare disorders, globally.
⏰Secure your place by 11 March: bit.ly/3BpAe44

February 17, 2025 at 1:22 PM
Join leading experts working in #RareDisease research at our #GRD25 conference.
📅 Dates: 9-11 April 2025
💭 Share insights in person
Explore the latest #genomics advances accelerating improvements in clinical care for rare disorders, globally.
⏰Secure your place by 11 March: bit.ly/3BpAe44
📅 Dates: 9-11 April 2025
💭 Share insights in person
Explore the latest #genomics advances accelerating improvements in clinical care for rare disorders, globally.
⏰Secure your place by 11 March: bit.ly/3BpAe44
Recently out on #bioRxiv: our updated approach to identify regional variability in missense mutation intolerance (“constraint”) in protein-coding genes using the gnomAD database.
www.biorxiv.org/content/10.1...
1/10
www.biorxiv.org/content/10.1...
1/10
April 19, 2024 at 11:34 PM
Recently out on #bioRxiv: our updated approach to identify regional variability in missense mutation intolerance (“constraint”) in protein-coding genes using the gnomAD database.
www.biorxiv.org/content/10.1...
1/10
www.biorxiv.org/content/10.1...
1/10
Some updated guidance on our gnomAD v4 constraint scores: gnomad.broadinstitute.org/news/2024-03...
The @gnomad-project.bsky.social team is hard at work on v4.1 and improvements across the board, so expect more updates.
Thanks to Katherine Chao for spearheading this blogpost.
The @gnomad-project.bsky.social team is hard at work on v4.1 and improvements across the board, so expect more updates.
Thanks to Katherine Chao for spearheading this blogpost.
March 8, 2024 at 4:34 PM
Some updated guidance on our gnomAD v4 constraint scores: gnomad.broadinstitute.org/news/2024-03...
The @gnomad-project.bsky.social team is hard at work on v4.1 and improvements across the board, so expect more updates.
Thanks to Katherine Chao for spearheading this blogpost.
The @gnomad-project.bsky.social team is hard at work on v4.1 and improvements across the board, so expect more updates.
Thanks to Katherine Chao for spearheading this blogpost.
Our paper describing a way to infer the phase of rare variant pairs using gnomAD v2 is out now in Nature Genetics.
We hope that the resource we generated will be useful when interpreting rare co-occurring variants in the context of recessive disease.
www.nature.com/articles/s41...
We hope that the resource we generated will be useful when interpreting rare co-occurring variants in the context of recessive disease.
www.nature.com/articles/s41...
December 8, 2023 at 2:31 AM
Our paper describing a way to infer the phase of rare variant pairs using gnomAD v2 is out now in Nature Genetics.
We hope that the resource we generated will be useful when interpreting rare co-occurring variants in the context of recessive disease.
www.nature.com/articles/s41...
We hope that the resource we generated will be useful when interpreting rare co-occurring variants in the context of recessive disease.
www.nature.com/articles/s41...
It’s the final day of giving thanks for the teams that make gnomAD possible. Today is focused on the participants in studies, data contributors (>300!), the Scientific Advisory Board, and our steering committee.
1/6
1/6
November 17, 2023 at 6:59 PM
It’s the final day of giving thanks for the teams that make gnomAD possible. Today is focused on the participants in studies, data contributors (>300!), the Scientific Advisory Board, and our steering committee.
1/6
1/6
Day four of giving thanks to the teams that make gnomAD happen is focused on the CNV and SV teams!
v4 is the first time we released structural variants at the same time as SNVs/indels, specifically:
- CNVs from 464,297 exomes
- SVs from 63,046 genomes
1/4
v4 is the first time we released structural variants at the same time as SNVs/indels, specifically:
- CNVs from 464,297 exomes
- SVs from 63,046 genomes
1/4

November 16, 2023 at 9:36 PM
Day four of giving thanks to the teams that make gnomAD happen is focused on the CNV and SV teams!
v4 is the first time we released structural variants at the same time as SNVs/indels, specifically:
- CNVs from 464,297 exomes
- SVs from 63,046 genomes
1/4
v4 is the first time we released structural variants at the same time as SNVs/indels, specifically:
- CNVs from 464,297 exomes
- SVs from 63,046 genomes
1/4
Continuing the week of thanking the teams that make gnomAD possible, today I’m thanking the data generation and operations teams!
As a reminder, I'm only highlighting a few individuals of the many that contribute. You can see more on our team page:
gnomad.broadinstitute.org/team
1/5
As a reminder, I'm only highlighting a few individuals of the many that contribute. You can see more on our team page:
gnomad.broadinstitute.org/team
1/5

November 15, 2023 at 7:19 PM
Continuing the week of thanking the teams that make gnomAD possible, today I’m thanking the data generation and operations teams!
As a reminder, I'm only highlighting a few individuals of the many that contribute. You can see more on our team page:
gnomad.broadinstitute.org/team
1/5
As a reminder, I'm only highlighting a few individuals of the many that contribute. You can see more on our team page:
gnomad.broadinstitute.org/team
1/5
Next up in my week of thanks to the teams that make gnomAD: the gnomAD browser team!
With 150k+ views a week, the browser is a crucial part of making gnomAD accessible. Quickly loading data from >800k samples + presenting it in a user-friendly format is no small feat.
1/6
With 150k+ views a week, the browser is a crucial part of making gnomAD accessible. Quickly loading data from >800k samples + presenting it in a user-friendly format is no small feat.
1/6

November 14, 2023 at 8:04 PM
Next up in my week of thanks to the teams that make gnomAD: the gnomAD browser team!
With 150k+ views a week, the browser is a crucial part of making gnomAD accessible. Quickly loading data from >800k samples + presenting it in a user-friendly format is no small feat.
1/6
With 150k+ views a week, the browser is a crucial part of making gnomAD accessible. Quickly loading data from >800k samples + presenting it in a user-friendly format is no small feat.
1/6
Now that the dust has settled on the gnomAD v4 release, which hopefully many of you have already checked out, I wanted to take this week to thank many of the members of the team who made this possible.
First up this week is the amazing production team.
1/7
First up this week is the amazing production team.
1/7

November 13, 2023 at 7:46 PM
Now that the dust has settled on the gnomAD v4 release, which hopefully many of you have already checked out, I wanted to take this week to thank many of the members of the team who made this possible.
First up this week is the amazing production team.
1/7
First up this week is the amazing production team.
1/7
Reposted by Kaitlin Samocha

Helpful description of the choices of population designators in gnomAD: gnomad.broadinstitute.org/news/2023-11...
November 9, 2023 at 4:01 PM
Helpful description of the choices of population designators in gnomAD: gnomad.broadinstitute.org/news/2023-11...
Constraint scores are now up for v4!

November 3, 2023 at 9:44 PM
Constraint scores are now up for v4!
Reposted by Kaitlin Samocha

To learn more about the impact of diversity on variant discovery and gene constraint please attend Katherine Chao’s #ASHG23 talk tomorrow (11/4) at 11am in rm 202A
Our genetic ancestry blog broad.io/gnomad_ancestry discusses our efforts to improve representation in #gnomAD, how we label groups and how the diversity in gnomAD is improving genomic filtration. (4/11)
November 3, 2023 at 1:01 PM
To learn more about the impact of diversity on variant discovery and gene constraint please attend Katherine Chao’s #ASHG23 talk tomorrow (11/4) at 11am in rm 202A
Reposted by Kaitlin Samocha
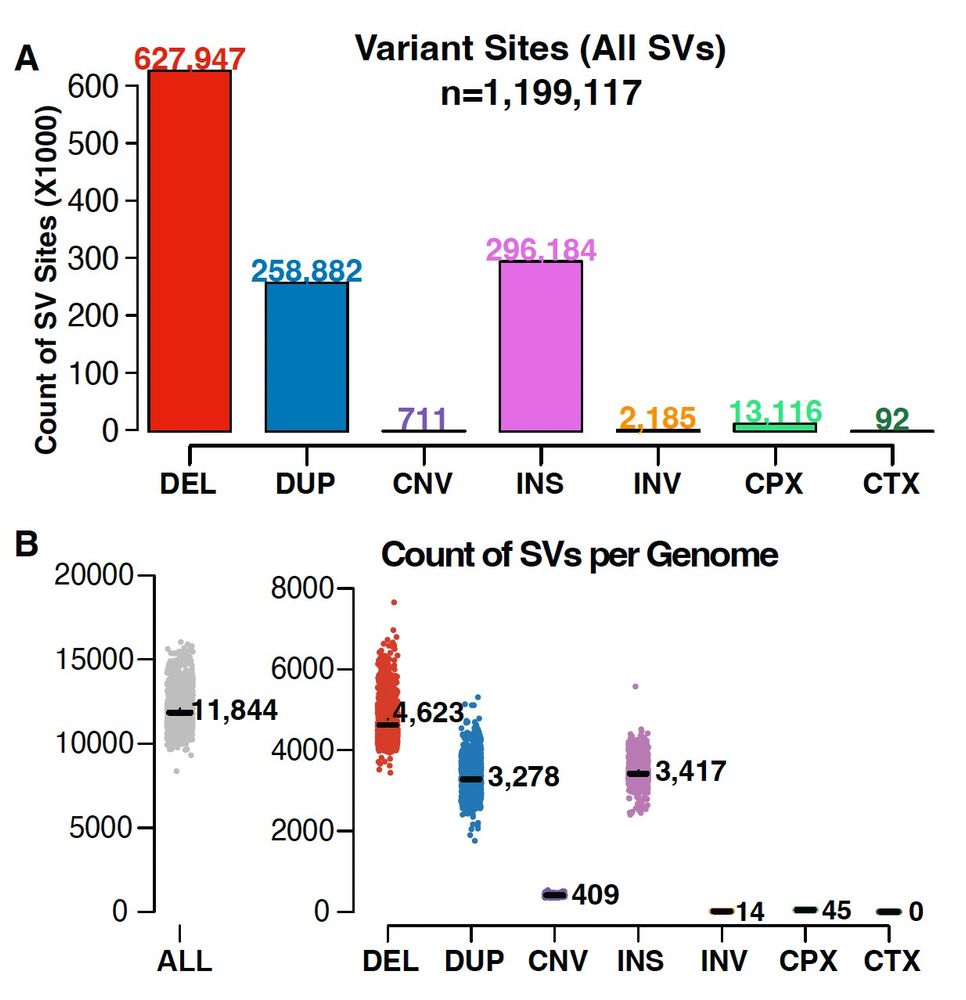
As part of #gnomAD v4, in collaboration with the Talkowski Lab, we have released 1,199,117 genome SVs and 66,903 rare exome CNVs. These data represent the first gnomAD SV dataset released native to the GRCh38 reference genome. (1/2)
November 2, 2023 at 12:58 PM
As part of #gnomAD v4, in collaboration with the Talkowski Lab, we have released 1,199,117 genome SVs and 66,903 rare exome CNVs. These data represent the first gnomAD SV dataset released native to the GRCh38 reference genome. (1/2)