



Kaitlin Samocha
@ksamocha.bsky.social
Assistant Investigator @ MGH / Broad / HMS. Focus on human genomics and modeling rare variation. She/her
If this work feels familiar, it is because it is building off older work from our team originally released for ExAC.
I view this iteration as more of a franchise reboot instead of a sequel – we have new leads, but similar themes.
9/10
I view this iteration as more of a franchise reboot instead of a sequel – we have new leads, but similar themes.
9/10


April 19, 2024 at 11:39 PM
If this work feels familiar, it is because it is building off older work from our team originally released for ExAC.
I view this iteration as more of a franchise reboot instead of a sequel – we have new leads, but similar themes.
9/10
I view this iteration as more of a franchise reboot instead of a sequel – we have new leads, but similar themes.
9/10
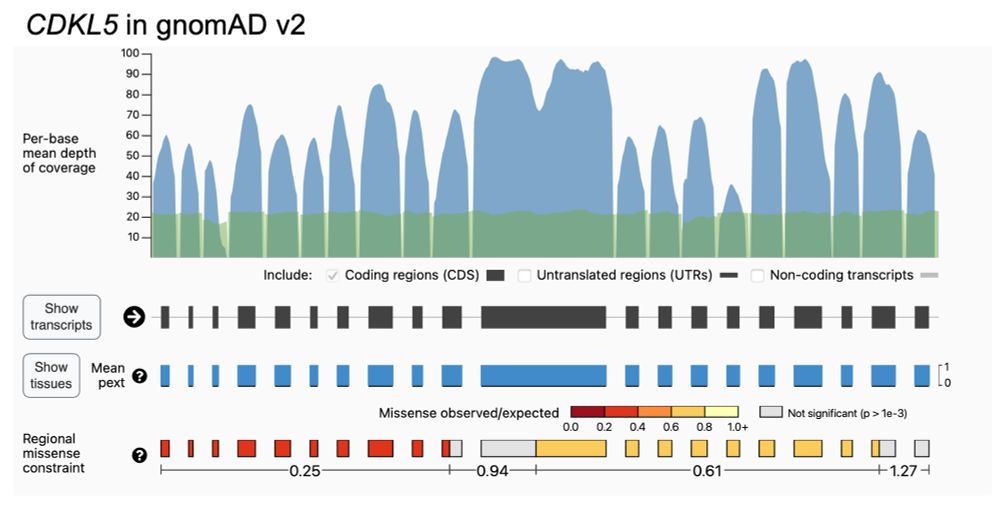
As with all gnomAD-led projects, we’ve already shared the data and code. Regions are displayed for v2 on the gnomAD browser, the code can be seen on Github (github.com/broadinstitu...), and MPC scores are available for download.
8/10
8/10
April 19, 2024 at 11:39 PM
As with all gnomAD-led projects, we’ve already shared the data and code. Regions are displayed for v2 on the gnomAD browser, the code can be seen on Github (github.com/broadinstitu...), and MPC scores are available for download.
8/10
8/10
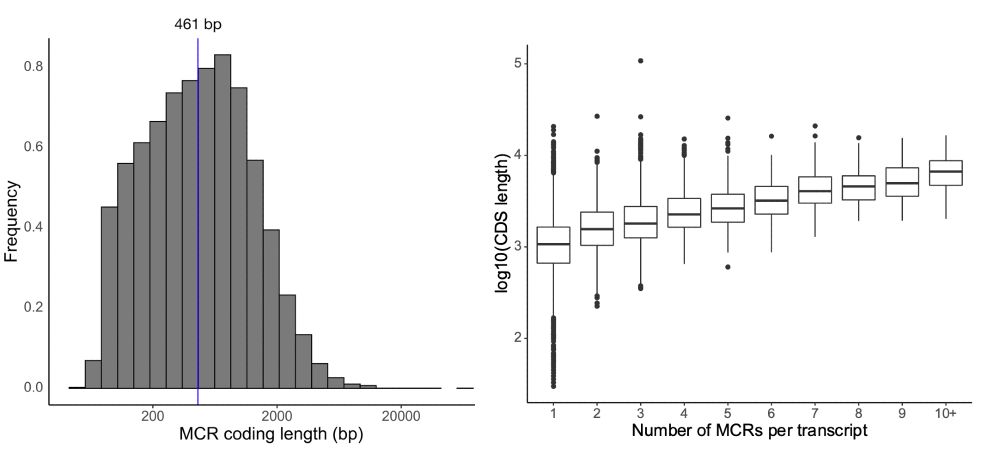
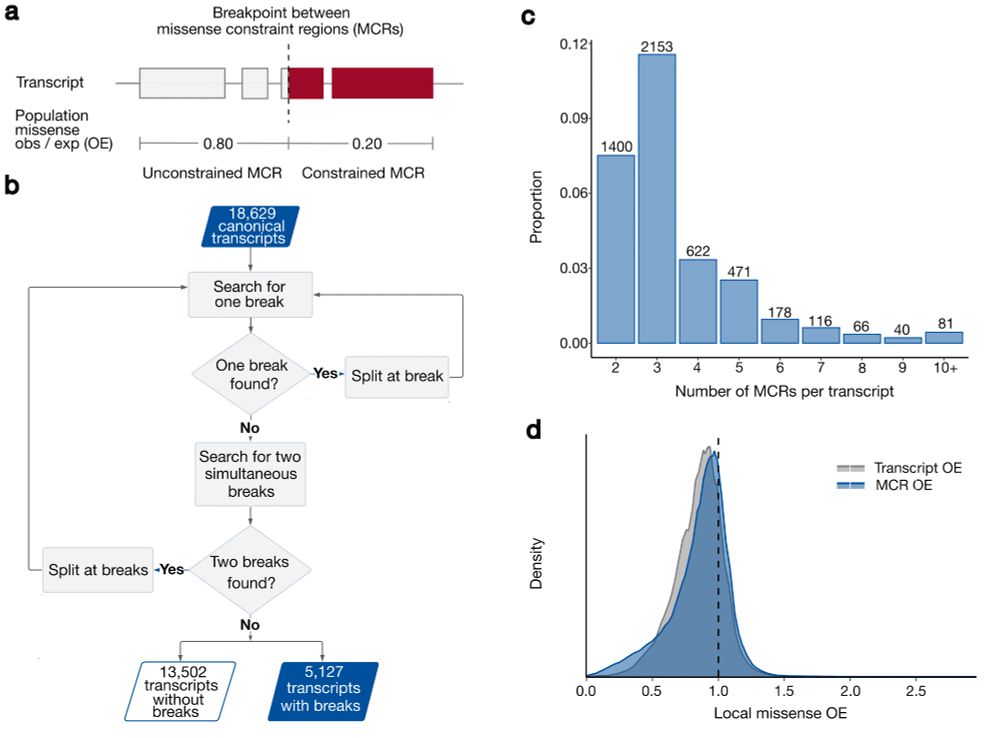
There is still much more to learn: using 125k exomes, our median region size is ~450bp and we see a relationship between transcript length and the number of regions we can identify due to statistical power.
7/10
7/10
April 19, 2024 at 11:38 PM
There is still much more to learn: using 125k exomes, our median region size is ~450bp and we see a relationship between transcript length and the number of regions we can identify due to statistical power.
7/10
7/10
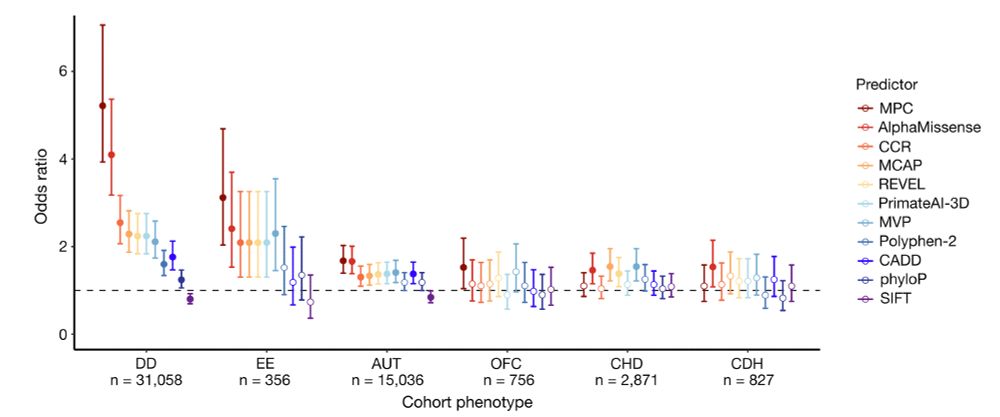
Finally, missense constraint information was incorporated into a deleteriousness metric named MPC (Missense deleteriousness Prediction by Constraint), which separates case from control de novo missense variants well with similar performance to ML models like AlphaMissense.
6/10
6/10
April 19, 2024 at 11:37 PM
Finally, missense constraint information was incorporated into a deleteriousness metric named MPC (Missense deleteriousness Prediction by Constraint), which separates case from control de novo missense variants well with similar performance to ML models like AlphaMissense.
6/10
6/10
In collaboration with Predrag Radivojac and team, we demonstrated that coding bases with < 20% of their expected missense variation achieve moderate support for pathogenicity (PM1) following ACMG/AMP guidelines that can be used for clinical classification.
5/10
5/10

April 19, 2024 at 11:37 PM
In collaboration with Predrag Radivojac and team, we demonstrated that coding bases with < 20% of their expected missense variation achieve moderate support for pathogenicity (PM1) following ACMG/AMP guidelines that can be used for clinical classification.
5/10
5/10
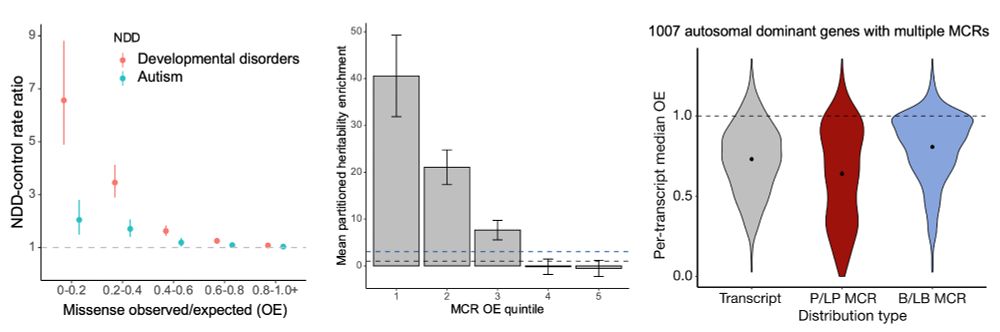
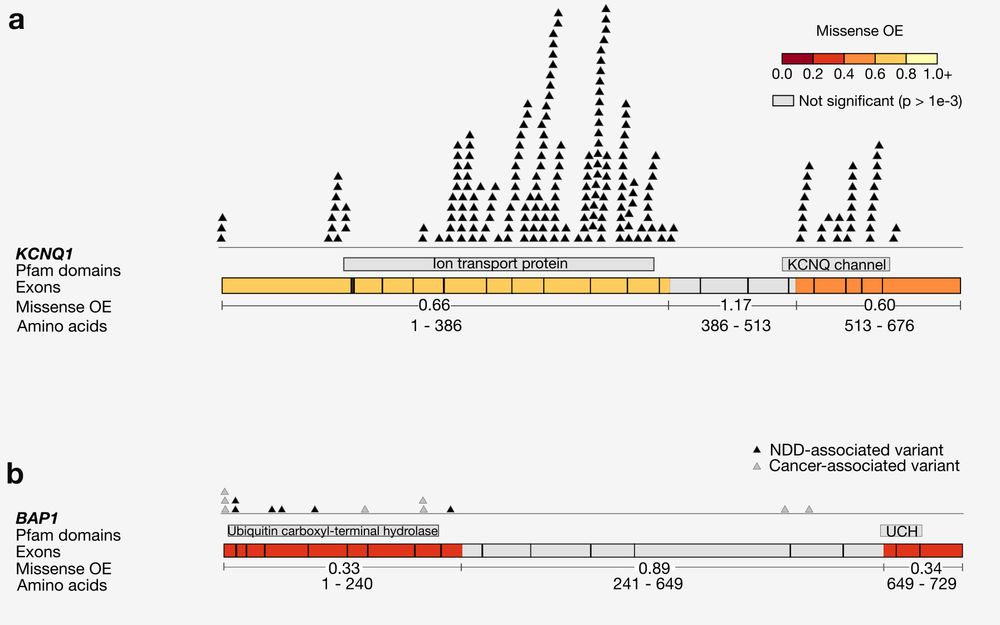
Missense depleted regions show an enrichment of (1) de novo missense variants in neurodevelopmental disorder cases compared to controls, (2) partitioned common variant heritability for >260 independent traits from the UK Biobank, and (3) ClinVar pathogenic (P/LP) variants.
4/10
4/10
April 19, 2024 at 11:36 PM
Missense depleted regions show an enrichment of (1) de novo missense variants in neurodevelopmental disorder cases compared to controls, (2) partitioned common variant heritability for >260 independent traits from the UK Biobank, and (3) ClinVar pathogenic (P/LP) variants.
4/10
4/10
Why try to find subgenic regions that are specifically missense constrained? Splitting up genes reveals patterns of negative and neutral selection that are obscured when looking gene-wide, including highlighting regions that have a large number of known pathogenic variants.
3/10
3/10
April 19, 2024 at 11:35 PM
Why try to find subgenic regions that are specifically missense constrained? Splitting up genes reveals patterns of negative and neutral selection that are obscured when looking gene-wide, including highlighting regions that have a large number of known pathogenic variants.
3/10
3/10
Co-led by the fabulous Katherine Chao and Lily Wang, we used gnomAD v2 and a recursive search to identify ~28% of canonical transcripts that were split into multiple missense constraint regions (measured by variable missense depletion in gnomAD).
2/10
2/10
April 19, 2024 at 11:35 PM
Co-led by the fabulous Katherine Chao and Lily Wang, we used gnomAD v2 and a recursive search to identify ~28% of canonical transcripts that were split into multiple missense constraint regions (measured by variable missense depletion in gnomAD).
2/10
2/10
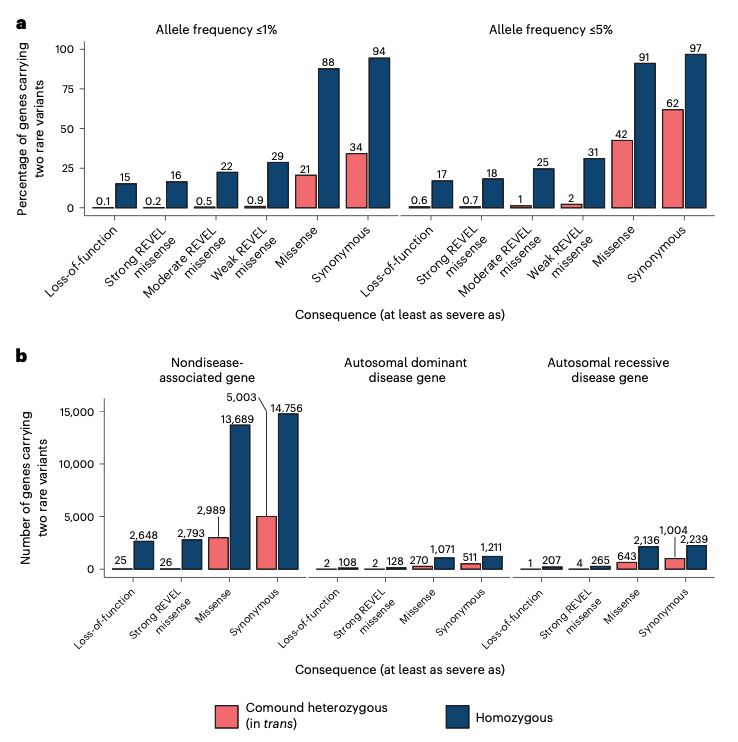
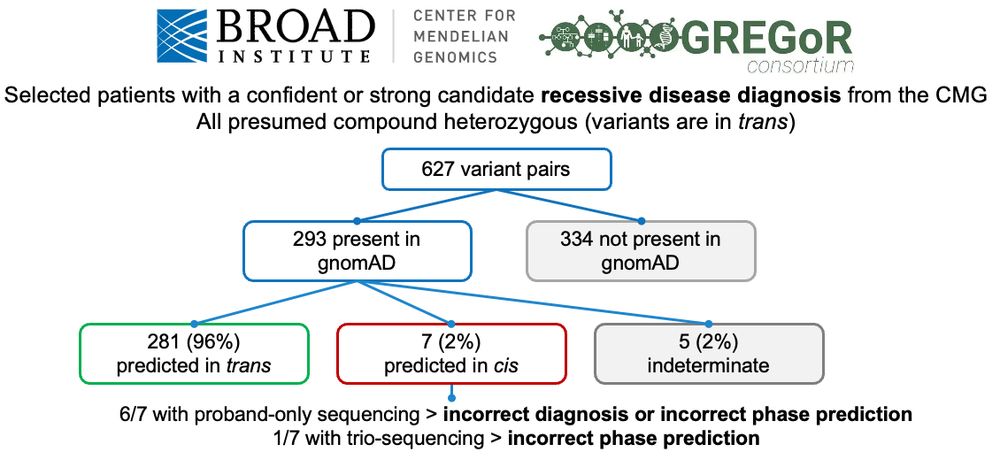
We also counted the number of genes that appeared to have compound heterozygous (in trans) deleterious variants and didn’t find that many. After manual curation, we only found seven genes with high-confidence compound heterozygous loss-of-function variants with some caveats.
December 8, 2023 at 2:36 AM
We also counted the number of genes that appeared to have compound heterozygous (in trans) deleterious variants and didn’t find that many. After manual curation, we only found seven genes with high-confidence compound heterozygous loss-of-function variants with some caveats.
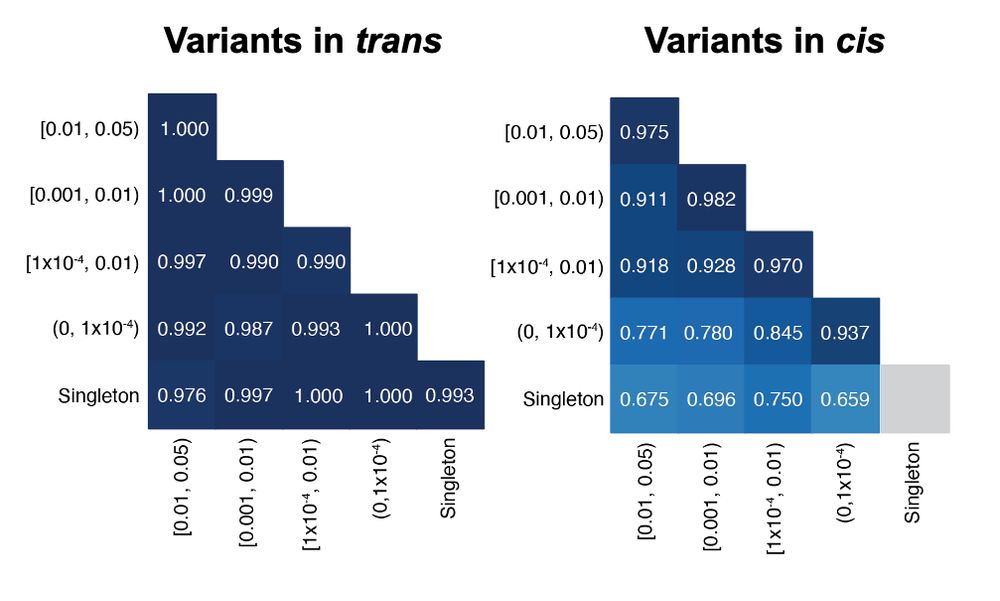
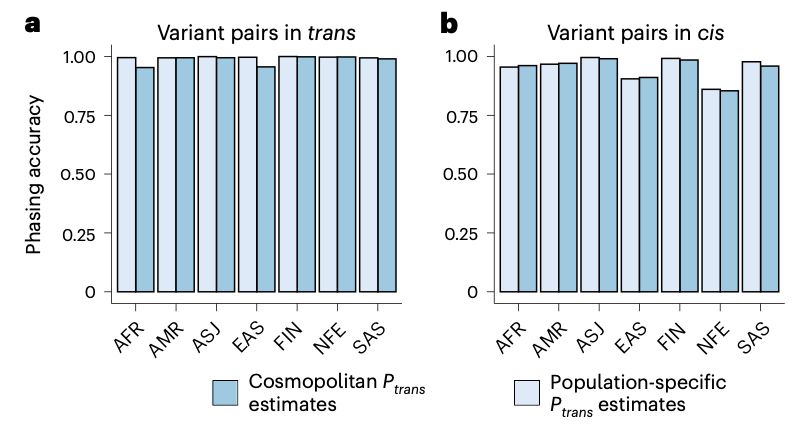
Accuracy of our approach remains high across a range of allele frequencies and across genetic ancestry groups. As expected, genetic distance and highly mutable sites both impact accuracy.
December 8, 2023 at 2:34 AM
Accuracy of our approach remains high across a range of allele frequencies and across genetic ancestry groups. As expected, genetic distance and highly mutable sites both impact accuracy.
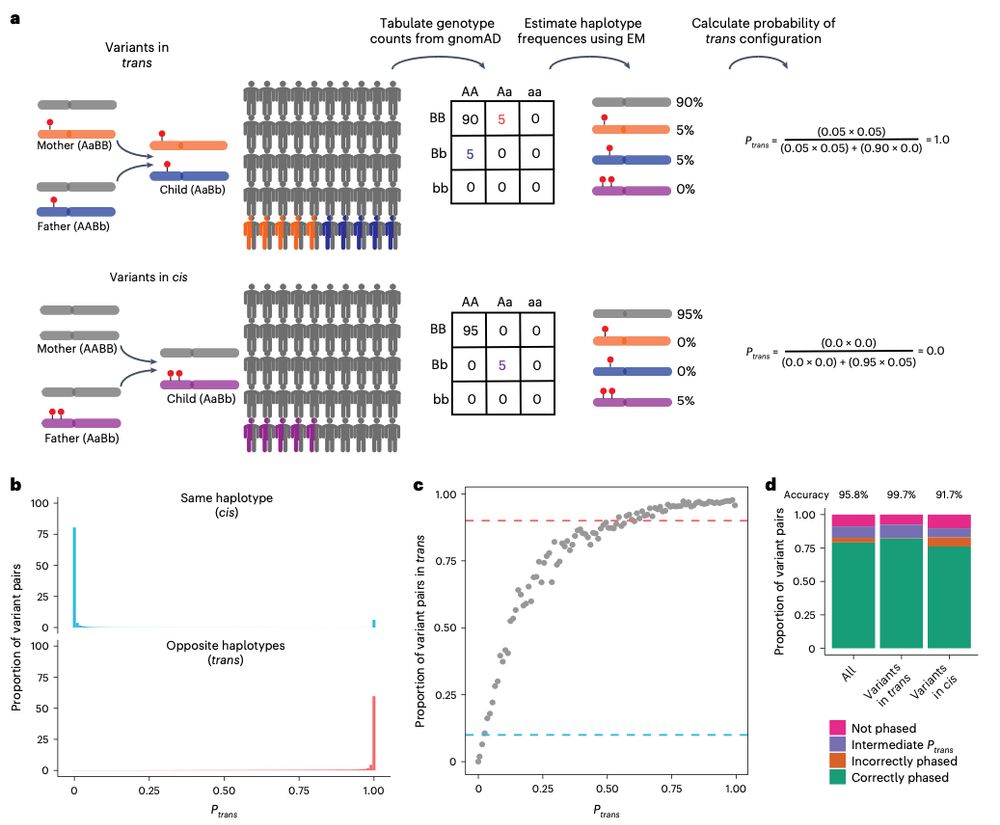
In this work, we used gnomAD v2 to estimate haplotype frequencies, and thereby predict the probability that two rare variants observed in the same gene are in trans. This approach is highly accurate (~96%) for variants with known phase in two independent datasets.
December 8, 2023 at 2:34 AM
In this work, we used gnomAD v2 to estimate haplotype frequencies, and thereby predict the probability that two rare variants observed in the same gene are in trans. This approach is highly accurate (~96%) for variants with known phase in two independent datasets.
Finally, I’d like to thank the steering committee of gnomAD, and particularly to our co-Directors: Mark Daly and Heidi Rehm. It’s been fun to be a part of this amazing project and process.
5/6
5/6

November 17, 2023 at 7:00 PM
Finally, I’d like to thank the steering committee of gnomAD, and particularly to our co-Directors: Mark Daly and Heidi Rehm. It’s been fun to be a part of this amazing project and process.
5/6
5/6
On the leadership side, we have a wonderful Scientific Advisory Board who provides suggestions and guidance for us. I’m looking forward to more discussions about how we can make this resource better.
4/6
4/6

November 17, 2023 at 7:00 PM
On the leadership side, we have a wonderful Scientific Advisory Board who provides suggestions and guidance for us. I’m looking forward to more discussions about how we can make this resource better.
4/6
4/6
Of course, gnomAD isn’t a reference population resource without the individuals who agreed to participate in research and shared their genetic information.
To all of you: thank you for contributing. You are the heart of gnomAD.
2/6
To all of you: thank you for contributing. You are the heart of gnomAD.
2/6

November 17, 2023 at 6:59 PM
Of course, gnomAD isn’t a reference population resource without the individuals who agreed to participate in research and shared their genetic information.
To all of you: thank you for contributing. You are the heart of gnomAD.
2/6
To all of you: thank you for contributing. You are the heart of gnomAD.
2/6
Day four of giving thanks to the teams that make gnomAD happen is focused on the CNV and SV teams!
v4 is the first time we released structural variants at the same time as SNVs/indels, specifically:
- CNVs from 464,297 exomes
- SVs from 63,046 genomes
1/4
v4 is the first time we released structural variants at the same time as SNVs/indels, specifically:
- CNVs from 464,297 exomes
- SVs from 63,046 genomes
1/4

November 16, 2023 at 9:36 PM
Day four of giving thanks to the teams that make gnomAD happen is focused on the CNV and SV teams!
v4 is the first time we released structural variants at the same time as SNVs/indels, specifically:
- CNVs from 464,297 exomes
- SVs from 63,046 genomes
1/4
v4 is the first time we released structural variants at the same time as SNVs/indels, specifically:
- CNVs from 464,297 exomes
- SVs from 63,046 genomes
1/4
Continuing the week of thanking the teams that make gnomAD possible, today I’m thanking the data generation and operations teams!
As a reminder, I'm only highlighting a few individuals of the many that contribute. You can see more on our team page:
gnomad.broadinstitute.org/team
1/5
As a reminder, I'm only highlighting a few individuals of the many that contribute. You can see more on our team page:
gnomad.broadinstitute.org/team
1/5

November 15, 2023 at 7:19 PM
Continuing the week of thanking the teams that make gnomAD possible, today I’m thanking the data generation and operations teams!
As a reminder, I'm only highlighting a few individuals of the many that contribute. You can see more on our team page:
gnomad.broadinstitute.org/team
1/5
As a reminder, I'm only highlighting a few individuals of the many that contribute. You can see more on our team page:
gnomad.broadinstitute.org/team
1/5
Elissa Alarmani is doing a co-op with our team and has already made valuable contributions. Specifically, she built the display for the exome CNVs in the last few months!! Definitely one to watch. 👀
6/6
6/6

November 14, 2023 at 8:06 PM
Elissa Alarmani is doing a co-op with our team and has already made valuable contributions. Specifically, she built the display for the exome CNVs in the last few months!! Definitely one to watch. 👀
6/6
6/6
Riley Grant was the point person for making sure all of the changes made to the FAQ were uploaded correctly. He also developed one of my favorite features (I’m biased) – the regional missense constraint track!
5/6
5/6

November 14, 2023 at 8:05 PM
Riley Grant was the point person for making sure all of the changes made to the FAQ were uploaded correctly. He also developed one of my favorite features (I’m biased) – the regional missense constraint track!
5/6
5/6
Phil Darnowsky was responsible for the display of structural variants (SVs) released for ~63k genome samples, which you can read more about here:
gnomad.broadinstitute.org/news/2023-11...
He also recently added the variant co-occurrence counts table to the gene pages in v2.
4/6
gnomad.broadinstitute.org/news/2023-11...
He also recently added the variant co-occurrence counts table to the gene pages in v2.
4/6

November 14, 2023 at 8:05 PM
Phil Darnowsky was responsible for the display of structural variants (SVs) released for ~63k genome samples, which you can read more about here:
gnomad.broadinstitute.org/news/2023-11...
He also recently added the variant co-occurrence counts table to the gene pages in v2.
4/6
gnomad.broadinstitute.org/news/2023-11...
He also recently added the variant co-occurrence counts table to the gene pages in v2.
4/6
Next up in my week of thanks to the teams that make gnomAD: the gnomAD browser team!
With 150k+ views a week, the browser is a crucial part of making gnomAD accessible. Quickly loading data from >800k samples + presenting it in a user-friendly format is no small feat.
1/6
With 150k+ views a week, the browser is a crucial part of making gnomAD accessible. Quickly loading data from >800k samples + presenting it in a user-friendly format is no small feat.
1/6

November 14, 2023 at 8:04 PM
Next up in my week of thanks to the teams that make gnomAD: the gnomAD browser team!
With 150k+ views a week, the browser is a crucial part of making gnomAD accessible. Quickly loading data from >800k samples + presenting it in a user-friendly format is no small feat.
1/6
With 150k+ views a week, the browser is a crucial part of making gnomAD accessible. Quickly loading data from >800k samples + presenting it in a user-friendly format is no small feat.
1/6
Now that the dust has settled on the gnomAD v4 release, which hopefully many of you have already checked out, I wanted to take this week to thank many of the members of the team who made this possible.
First up this week is the amazing production team.
1/7
First up this week is the amazing production team.
1/7

November 13, 2023 at 7:46 PM
Now that the dust has settled on the gnomAD v4 release, which hopefully many of you have already checked out, I wanted to take this week to thank many of the members of the team who made this possible.
First up this week is the amazing production team.
1/7
First up this week is the amazing production team.
1/7
Constraint scores are now up for v4!

November 3, 2023 at 9:44 PM
Constraint scores are now up for v4!