



Kaitlin Samocha
@ksamocha.bsky.social
Assistant Investigator @ MGH / Broad / HMS. Focus on human genomics and modeling rare variation. She/her
I haven't found it printed but there is a PDF link here: www.ashg.org/wp-content/u...
www.ashg.org
October 16, 2025 at 9:17 PM
I haven't found it printed but there is a PDF link here: www.ashg.org/wp-content/u...
Yes, I've done this once before for a paper (listing in the system only first and last authors). It is easy to get those handful of people to approve additional authors in the future if the journal requires it.
Not sure if recommended, but it saved time.
Not sure if recommended, but it saved time.
February 21, 2025 at 5:33 PM
Yes, I've done this once before for a paper (listing in the system only first and last authors). It is easy to get those handful of people to approve additional authors in the future if the journal requires it.
Not sure if recommended, but it saved time.
Not sure if recommended, but it saved time.
Projects like this can’t be completed without many others: thanks to Mark Daly for continued mentorship across the years; critical support and work from @anneotation.bsky.social, @konradjk.bsky.social, Predrag Radivojac; the Hail team; and everyone associated with @gnomad-project.bsky.social.
10/10
10/10
April 19, 2024 at 11:42 PM
Projects like this can’t be completed without many others: thanks to Mark Daly for continued mentorship across the years; critical support and work from @anneotation.bsky.social, @konradjk.bsky.social, Predrag Radivojac; the Hail team; and everyone associated with @gnomad-project.bsky.social.
10/10
10/10
If this work feels familiar, it is because it is building off older work from our team originally released for ExAC.
I view this iteration as more of a franchise reboot instead of a sequel – we have new leads, but similar themes.
9/10
I view this iteration as more of a franchise reboot instead of a sequel – we have new leads, but similar themes.
9/10


April 19, 2024 at 11:39 PM
If this work feels familiar, it is because it is building off older work from our team originally released for ExAC.
I view this iteration as more of a franchise reboot instead of a sequel – we have new leads, but similar themes.
9/10
I view this iteration as more of a franchise reboot instead of a sequel – we have new leads, but similar themes.
9/10
As with all gnomAD-led projects, we’ve already shared the data and code. Regions are displayed for v2 on the gnomAD browser, the code can be seen on Github (github.com/broadinstitu...), and MPC scores are available for download.
8/10
8/10
April 19, 2024 at 11:39 PM
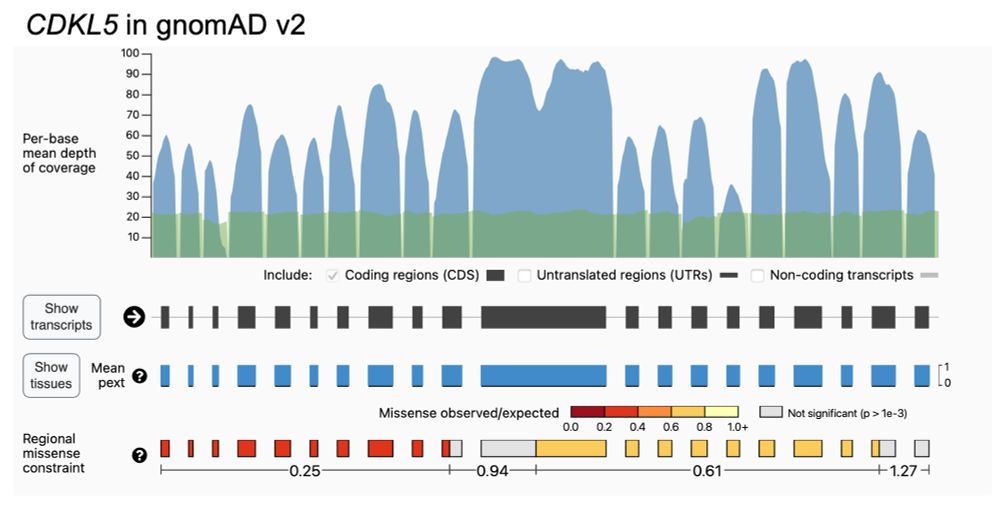
As with all gnomAD-led projects, we’ve already shared the data and code. Regions are displayed for v2 on the gnomAD browser, the code can be seen on Github (github.com/broadinstitu...), and MPC scores are available for download.
8/10
8/10
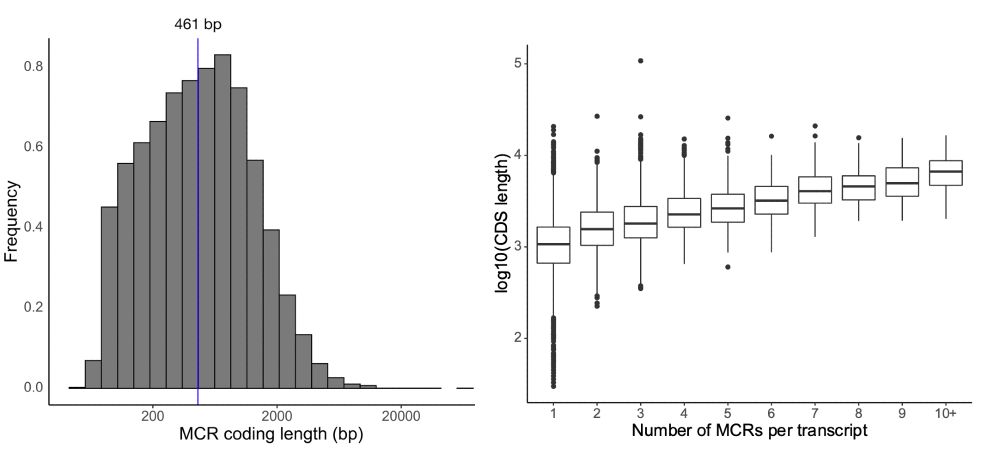
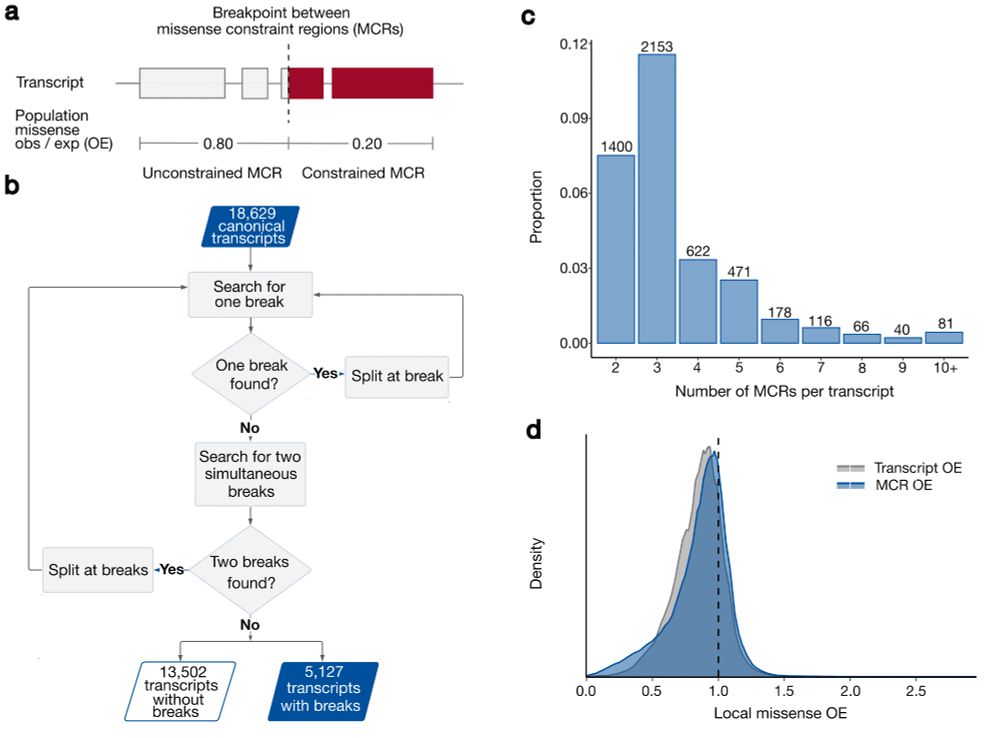
There is still much more to learn: using 125k exomes, our median region size is ~450bp and we see a relationship between transcript length and the number of regions we can identify due to statistical power.
7/10
7/10
April 19, 2024 at 11:38 PM
There is still much more to learn: using 125k exomes, our median region size is ~450bp and we see a relationship between transcript length and the number of regions we can identify due to statistical power.
7/10
7/10
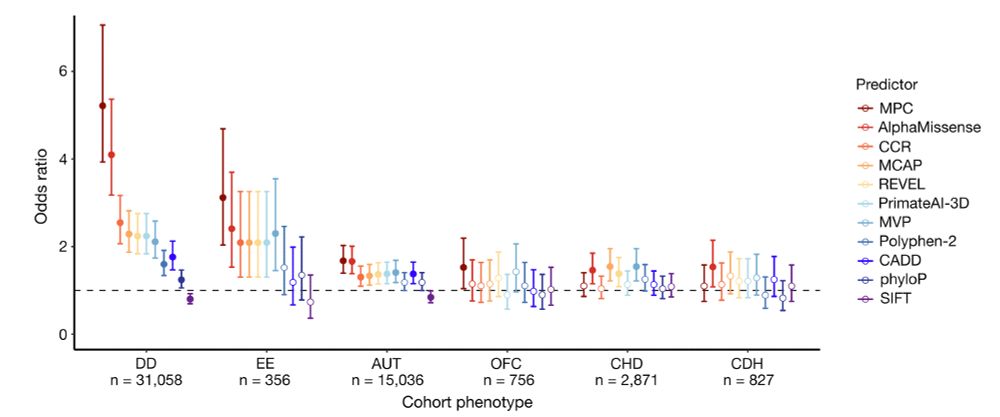
Finally, missense constraint information was incorporated into a deleteriousness metric named MPC (Missense deleteriousness Prediction by Constraint), which separates case from control de novo missense variants well with similar performance to ML models like AlphaMissense.
6/10
6/10
April 19, 2024 at 11:37 PM
Finally, missense constraint information was incorporated into a deleteriousness metric named MPC (Missense deleteriousness Prediction by Constraint), which separates case from control de novo missense variants well with similar performance to ML models like AlphaMissense.
6/10
6/10
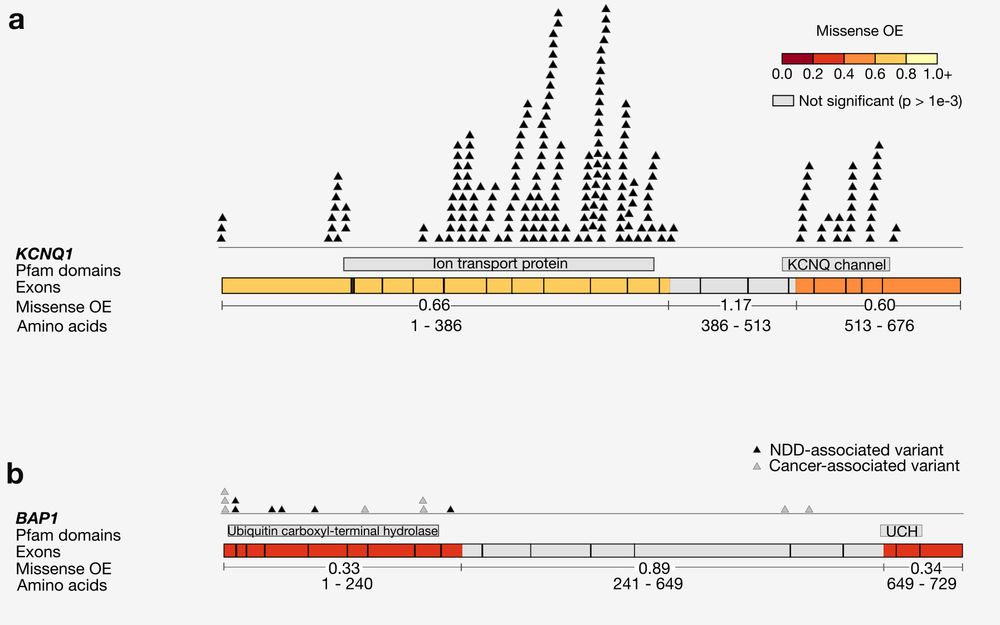
In collaboration with Predrag Radivojac and team, we demonstrated that coding bases with < 20% of their expected missense variation achieve moderate support for pathogenicity (PM1) following ACMG/AMP guidelines that can be used for clinical classification.
5/10
5/10

April 19, 2024 at 11:37 PM
In collaboration with Predrag Radivojac and team, we demonstrated that coding bases with < 20% of their expected missense variation achieve moderate support for pathogenicity (PM1) following ACMG/AMP guidelines that can be used for clinical classification.
5/10
5/10
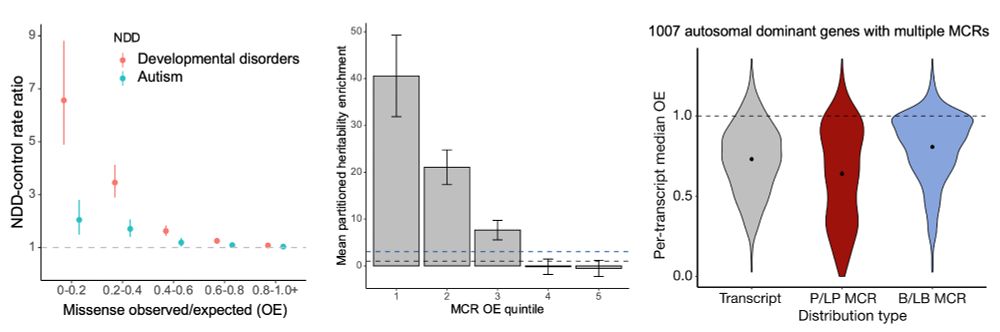
Missense depleted regions show an enrichment of (1) de novo missense variants in neurodevelopmental disorder cases compared to controls, (2) partitioned common variant heritability for >260 independent traits from the UK Biobank, and (3) ClinVar pathogenic (P/LP) variants.
4/10
4/10
April 19, 2024 at 11:36 PM
Missense depleted regions show an enrichment of (1) de novo missense variants in neurodevelopmental disorder cases compared to controls, (2) partitioned common variant heritability for >260 independent traits from the UK Biobank, and (3) ClinVar pathogenic (P/LP) variants.
4/10
4/10
Why try to find subgenic regions that are specifically missense constrained? Splitting up genes reveals patterns of negative and neutral selection that are obscured when looking gene-wide, including highlighting regions that have a large number of known pathogenic variants.
3/10
3/10
April 19, 2024 at 11:35 PM
Why try to find subgenic regions that are specifically missense constrained? Splitting up genes reveals patterns of negative and neutral selection that are obscured when looking gene-wide, including highlighting regions that have a large number of known pathogenic variants.
3/10
3/10
Co-led by the fabulous Katherine Chao and Lily Wang, we used gnomAD v2 and a recursive search to identify ~28% of canonical transcripts that were split into multiple missense constraint regions (measured by variable missense depletion in gnomAD).
2/10
2/10
April 19, 2024 at 11:35 PM
Co-led by the fabulous Katherine Chao and Lily Wang, we used gnomAD v2 and a recursive search to identify ~28% of canonical transcripts that were split into multiple missense constraint regions (measured by variable missense depletion in gnomAD).
2/10
2/10