



@countrsignal.bsky.social
Reposted

Structural biology is in an era of dynamics & assemblies but turning raw experimental data into atomic models at scale remains challenging. @minhuanli.bsky.social and I present ROCKET🚀: an AlphaFold augmentation that integrates crystallographic and cryoEM/ET data with room for more! 1/14.
February 24, 2025 at 12:23 PM
Structural biology is in an era of dynamics & assemblies but turning raw experimental data into atomic models at scale remains challenging. @minhuanli.bsky.social and I present ROCKET🚀: an AlphaFold augmentation that integrates crystallographic and cryoEM/ET data with room for more! 1/14.
Reposted

Can we achieve such an initial condition? There's a trick! We create synthetic plasmid dimers, transform them into cells. Then, we activate a recombinase that converts them back to monomers, ensuring an initial condition with equilibrated plasmid composition! (5/n)

February 21, 2025 at 8:40 PM
Can we achieve such an initial condition? There's a trick! We create synthetic plasmid dimers, transform them into cells. Then, we activate a recombinase that converts them back to monomers, ensuring an initial condition with equilibrated plasmid composition! (5/n)
Reposted
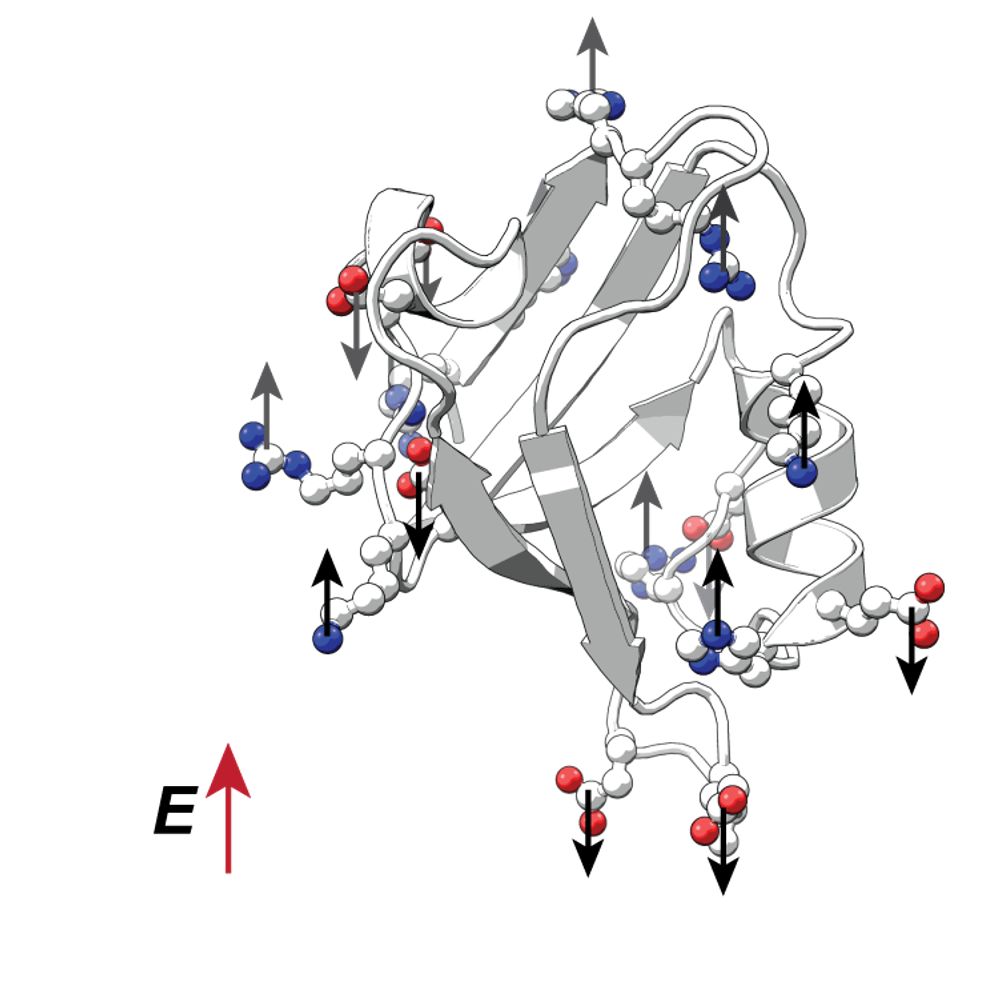
Crystal structures are *not* God-given truth. They approximate, w/ flaws & errors, X-ray diffraction data. AlphaFold etc. have been trained on structures, not data. SFCalculator now differentiably connects structures to diffraction data. What does this enable? 🧵 1/4 www.biorxiv.org/content/10.1...

February 23, 2025 at 1:52 AM
Crystal structures are *not* God-given truth. They approximate, w/ flaws & errors, X-ray diffraction data. AlphaFold etc. have been trained on structures, not data. SFCalculator now differentiably connects structures to diffraction data. What does this enable? 🧵 1/4 www.biorxiv.org/content/10.1...
Reposted

Acellera and Psivant Collaborate to Develop Transformative Computational Drug Discovery Approaches Using AI and Quantum Simulations
www.acellera.com/blog/aceller...
www.acellera.com/blog/aceller...

Acellera and Psivant Collaborate to Develop Transformative Computational Drug Discovery Approaches Using AI and Quantum Simulations - Acellera Blog
www.acellera.com
January 29, 2025 at 8:38 PM
Acellera and Psivant Collaborate to Develop Transformative Computational Drug Discovery Approaches Using AI and Quantum Simulations
www.acellera.com/blog/aceller...
www.acellera.com/blog/aceller...
Reposted
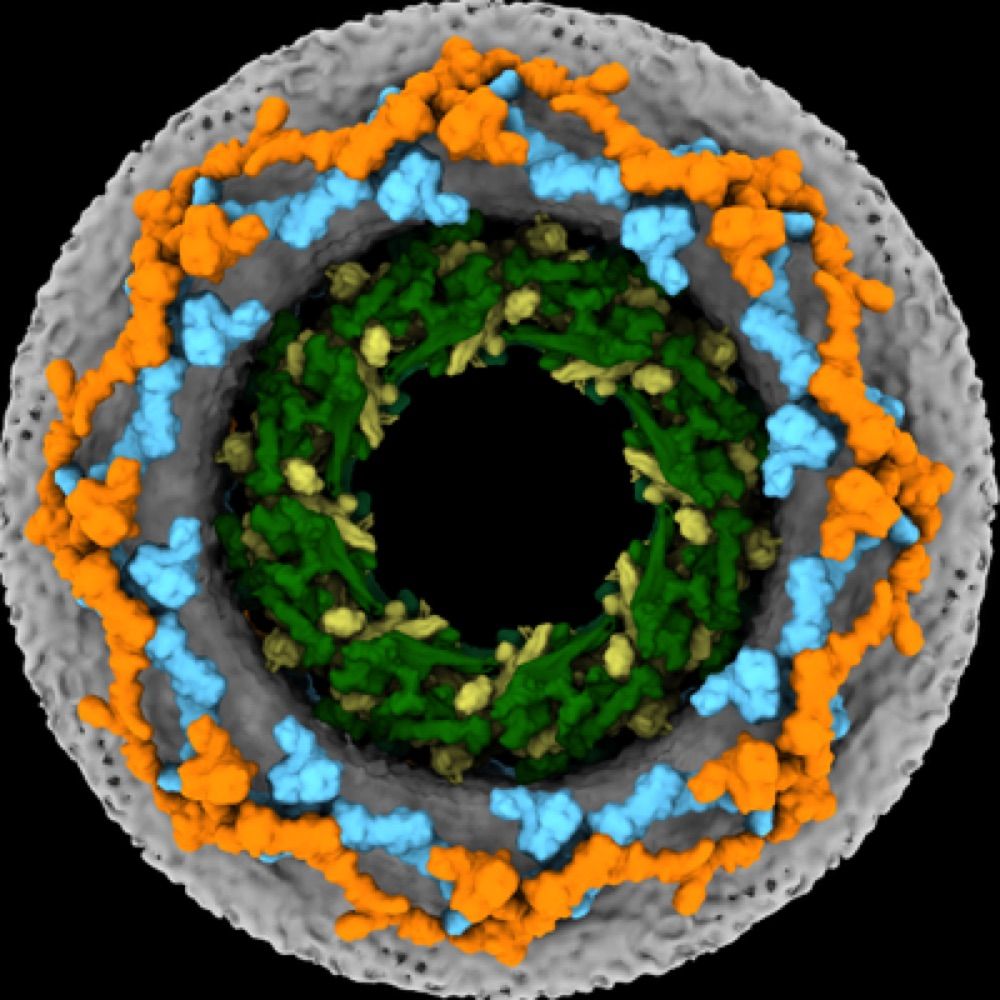
Through MD simulations we show that capsids face a significant steric barrier inside the NPC, especially when the vast amount of intrinsically disordered FG-Nups in the central channel are included. This barrier could be relieved by a crack in the NPC.
January 17, 2025 at 6:43 PM
Through MD simulations we show that capsids face a significant steric barrier inside the NPC, especially when the vast amount of intrinsically disordered FG-Nups in the central channel are included. This barrier could be relieved by a crack in the NPC.
Reposted
In a great collaboration with @hummerlab.bsky.social and the Kräusslich lab: HIV capsid doesn't break at the NPC; instead, it cracks open the NPC itself! Details in Cell: authors.elsevier.com/sd/article/S... @mpibp.bsky.social @uniheidelberg.bsky.social A thread below:
January 17, 2025 at 6:43 PM
In a great collaboration with @hummerlab.bsky.social and the Kräusslich lab: HIV capsid doesn't break at the NPC; instead, it cracks open the NPC itself! Details in Cell: authors.elsevier.com/sd/article/S... @mpibp.bsky.social @uniheidelberg.bsky.social A thread below:
Reposted

Ending the year with the nice closure of finally getting a long running project published. doi.org/10.1021/acs....

Accurate Estimation of Diffusion Coefficients and their Uncertainties from Computer Simulation
Self-diffusion coefficients, D*, are routinely estimated from molecular dynamics simulations by fitting a linear model to the observed mean squared displacements (MSDs) of mobile species. MSDs derived from simulations exhibit statistical noise that causes uncertainty in the resulting estimate of D*. An optimal scheme for estimating D* minimizes this uncertainty, i.e., it will have high statistical efficiency, and also gives an accurate estimate of the uncertainty itself. We present a scheme for estimating D* from a single simulation trajectory with a high statistical efficiency and accurately estimating the uncertainty in the predicted value. The statistical distribution of MSDs observable from a given simulation is modeled as a multivariate normal distribution using an analytical covariance matrix for an equivalent system of freely diffusing particles, which we parametrize from the available simulation data. We use Bayesian regression to sample the distribution of linear models that are compatible with this multivariate normal distribution to obtain a statistically efficient estimate of D* and an accurate estimate of the associated statistical uncertainty.
doi.org
December 30, 2024 at 9:17 PM
Ending the year with the nice closure of finally getting a long running project published. doi.org/10.1021/acs....
Reposted

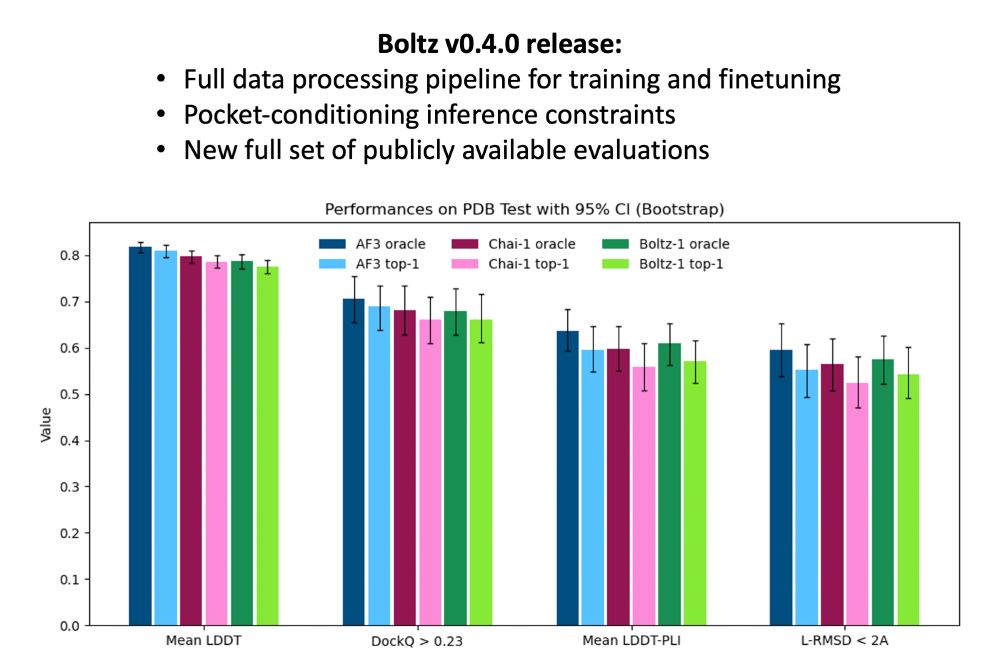
Boltz v0.4.0 is here! Today, we’re releasing our full data processing pipeline, making it easier than ever to build on top of Boltz. This release also includes our evaluation code and new results. Oh, and also pocket conditioning :)
December 21, 2024 at 4:26 PM
Boltz v0.4.0 is here! Today, we’re releasing our full data processing pipeline, making it easier than ever to build on top of Boltz. This release also includes our evaluation code and new results. Oh, and also pocket conditioning :)
Reposted

In biomolecular simulations, interesting phenomena often unfold slowly, with the viscosity of the surrounding water further hindering conformational sampling.
But what if we could simulate water with lower viscosity while preserving the same thermodynamic properties?
👇
But what if we could simulate water with lower viscosity while preserving the same thermodynamic properties?
👇
December 16, 2024 at 3:12 AM
In biomolecular simulations, interesting phenomena often unfold slowly, with the viscosity of the surrounding water further hindering conformational sampling.
But what if we could simulate water with lower viscosity while preserving the same thermodynamic properties?
👇
But what if we could simulate water with lower viscosity while preserving the same thermodynamic properties?
👇
Reposted

Predicting absolute protein folding stability using generative models
@mcagiada.bsky.social @sokrypton.org & I used ESM-IF to predict ∆G for folding & conformational change
Paper, code and colab
📜 dx.doi.org/10.1002/pro....
💾 github.com/KULL-Centre/...
👩💻 colab.research.google.com/github/KULL-...
@mcagiada.bsky.social @sokrypton.org & I used ESM-IF to predict ∆G for folding & conformational change
Paper, code and colab
📜 dx.doi.org/10.1002/pro....
💾 github.com/KULL-Centre/...
👩💻 colab.research.google.com/github/KULL-...
December 14, 2024 at 2:58 PM
Predicting absolute protein folding stability using generative models
@mcagiada.bsky.social @sokrypton.org & I used ESM-IF to predict ∆G for folding & conformational change
Paper, code and colab
📜 dx.doi.org/10.1002/pro....
💾 github.com/KULL-Centre/...
👩💻 colab.research.google.com/github/KULL-...
@mcagiada.bsky.social @sokrypton.org & I used ESM-IF to predict ∆G for folding & conformational change
Paper, code and colab
📜 dx.doi.org/10.1002/pro....
💾 github.com/KULL-Centre/...
👩💻 colab.research.google.com/github/KULL-...
Reposted

1/🧬 Excited to share PLAID, our new approach for co-generating sequence and all-atom protein structures by sampling from the latent space of ESMFold. This requires only sequences during training, which unlocks more data and annotations:
bit.ly/plaid-proteins
🧵
bit.ly/plaid-proteins
🧵
December 6, 2024 at 5:44 PM
1/🧬 Excited to share PLAID, our new approach for co-generating sequence and all-atom protein structures by sampling from the latent space of ESMFold. This requires only sequences during training, which unlocks more data and annotations:
bit.ly/plaid-proteins
🧵
bit.ly/plaid-proteins
🧵
Reposted
You may have seen a recent pre-print [1] from Jain et al. with strongly worded claims against the experimental results in our DiffDock paper [2]. We initially declined to respond as we saw that this preprint contained falsehoods, misleading comparisons, seemingly deliberate omissions, ...1/n
December 8, 2024 at 9:37 PM
You may have seen a recent pre-print [1] from Jain et al. with strongly worded claims against the experimental results in our DiffDock paper [2]. We initially declined to respond as we saw that this preprint contained falsehoods, misleading comparisons, seemingly deliberate omissions, ...1/n
Reposted
Thank you @dkoes.compstruct.org, unfortunately, this paper from Jain et al. contains falsehoods, misleading comparisons, seemingly deliberate omissions, and is written in a tone not intended as a serious research paper. Please see our detailed response: www.linkedin.com/pulse/respon...
Response to Jain et al.
You may have seen a recent pre-print [1] from Jain et al. with strongly worded claims against the experimental results in our DiffDock paper [2].
www.linkedin.com
December 8, 2024 at 9:51 PM
Thank you @dkoes.compstruct.org, unfortunately, this paper from Jain et al. contains falsehoods, misleading comparisons, seemingly deliberate omissions, and is written in a tone not intended as a serious research paper. Please see our detailed response: www.linkedin.com/pulse/respon...
Reposted

Oh wow. Maybe kind of a harsh attack, but we really do need a healthy debate about docking benchmarks. While I'm excited to see this one being fought out, may I already suggest the next point of debate: can we find a better metric than the utterly inadequate 2 Å RMSD cutoff?
Been a while since I read a paper like this:
• "What [DiffDock] appears to be doing cannot be considered" docking
• "Results are ... contaminated with near neighbors to test cases"
• "Results for DiffDock were artifactual"
• "Results for other methods were incorrectly done"
arxiv.org/abs/2412.02889
• "What [DiffDock] appears to be doing cannot be considered" docking
• "Results are ... contaminated with near neighbors to test cases"
• "Results for DiffDock were artifactual"
• "Results for other methods were incorrectly done"
arxiv.org/abs/2412.02889

Deep-Learning Based Docking Methods: Fair Comparisons to Conventional Docking Workflows
The diffusion learning method, DiffDock, for docking small-molecule ligands into protein binding sites was recently introduced. Results included comparisons to more conventional docking approaches, wi...
arxiv.org
December 5, 2024 at 4:13 PM
Oh wow. Maybe kind of a harsh attack, but we really do need a healthy debate about docking benchmarks. While I'm excited to see this one being fought out, may I already suggest the next point of debate: can we find a better metric than the utterly inadequate 2 Å RMSD cutoff?
Reposted

Super excited to preprint our work on developing a Biomolecular Emulator (BioEmu): Scalable emulation of protein equilibrium ensembles with generative deep learning from @msftresearch.bsky.social ch AI for Science.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
December 6, 2024 at 8:39 AM
Super excited to preprint our work on developing a Biomolecular Emulator (BioEmu): Scalable emulation of protein equilibrium ensembles with generative deep learning from @msftresearch.bsky.social ch AI for Science.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
Reposted

December 5, 2024 at 12:30 PM
Reposted

Been a while since I read a paper like this:
• "What [DiffDock] appears to be doing cannot be considered" docking
• "Results are ... contaminated with near neighbors to test cases"
• "Results for DiffDock were artifactual"
• "Results for other methods were incorrectly done"
arxiv.org/abs/2412.02889
• "What [DiffDock] appears to be doing cannot be considered" docking
• "Results are ... contaminated with near neighbors to test cases"
• "Results for DiffDock were artifactual"
• "Results for other methods were incorrectly done"
arxiv.org/abs/2412.02889
Deep-Learning Based Docking Methods: Fair Comparisons to Conventional Docking Workflows
The diffusion learning method, DiffDock, for docking small-molecule ligands into protein binding sites was recently introduced. Results included comparisons to more conventional docking approaches, wi...
arxiv.org
December 5, 2024 at 3:36 PM
Been a while since I read a paper like this:
• "What [DiffDock] appears to be doing cannot be considered" docking
• "Results are ... contaminated with near neighbors to test cases"
• "Results for DiffDock were artifactual"
• "Results for other methods were incorrectly done"
arxiv.org/abs/2412.02889
• "What [DiffDock] appears to be doing cannot be considered" docking
• "Results are ... contaminated with near neighbors to test cases"
• "Results for DiffDock were artifactual"
• "Results for other methods were incorrectly done"
arxiv.org/abs/2412.02889
Reposted

We're looking forward to presenting this @workshopmlsb.bsky.social . This work was conceived and led by a group CPCB (@cmupittcompbio.bsky.social) students and learns a co-embedding of proteins and ligands to support ultra fast virtual screening. arxiv.org/pdf/2411.15418


November 26, 2024 at 1:28 PM
We're looking forward to presenting this @workshopmlsb.bsky.social . This work was conceived and led by a group CPCB (@cmupittcompbio.bsky.social) students and learns a co-embedding of proteins and ligands to support ultra fast virtual screening. arxiv.org/pdf/2411.15418
Reposted

Reposted

This paper by Seth Lloyd was the one that made me and my collaborators do the first work on quantum computing for chemistry, which is now a very active field. www.science.org/doi/10.1126/...

Universal Quantum Simulators
Feynman's 1982 conjecture, that quantum computers can be programmed to simulate any local quantum system, is shown to be correct.
www.science.org
November 27, 2024 at 11:56 AM
This paper by Seth Lloyd was the one that made me and my collaborators do the first work on quantum computing for chemistry, which is now a very active field. www.science.org/doi/10.1126/...
Reposted

Beta-lactoglobulin having a little wiggle. The main whey protein in the milk of many mammals (except humans). Seen here traversing various configurations determined using NMR (1dv9.pdb). Imagine it dancing to music of your choice😂.
🧪⚗️ 🧶🧬🐡 #c4d #CompChemSky #SciComm
🧪⚗️ 🧶🧬🐡 #c4d #CompChemSky #SciComm
November 28, 2024 at 11:41 AM
Beta-lactoglobulin having a little wiggle. The main whey protein in the milk of many mammals (except humans). Seen here traversing various configurations determined using NMR (1dv9.pdb). Imagine it dancing to music of your choice😂.
🧪⚗️ 🧶🧬🐡 #c4d #CompChemSky #SciComm
🧪⚗️ 🧶🧬🐡 #c4d #CompChemSky #SciComm
Reposted
We’re excited to release a major update to the Boltz repo: v0.3.0. This release contains several important new features, including our confidence model and memory-efficient inference. Give it a try! github.com/jwohlwend/bo...
GitHub - jwohlwend/boltz: Official repository for the Boltz-1 biomolecular interaction model
Official repository for the Boltz-1 biomolecular interaction model - jwohlwend/boltz
github.com
November 28, 2024 at 4:50 PM
We’re excited to release a major update to the Boltz repo: v0.3.0. This release contains several important new features, including our confidence model and memory-efficient inference. Give it a try! github.com/jwohlwend/bo...
Reposted

Recovering screenshots of stuff that should be on bluesky

November 26, 2024 at 10:01 PM
Recovering screenshots of stuff that should be on bluesky
Reposted
Here is how Boltz-1 (green), DynamicBind (magenta), and GNINA (blue) dock a collection of random molecules. GNINA, using a classical sampling algorithm (MCMC) hits all concave regions while the ML samplers have distinct preferences. Boltz is the most likely to induce a fit.
November 22, 2024 at 6:27 PM
Here is how Boltz-1 (green), DynamicBind (magenta), and GNINA (blue) dock a collection of random molecules. GNINA, using a classical sampling algorithm (MCMC) hits all concave regions while the ML samplers have distinct preferences. Boltz is the most likely to induce a fit.
Reposted

Today was a great day! Our paper on using an AI platform for cell-free RNA liquid biopsy is finally published in Nature Communications! Huge thanks to our amazing team at @exai.bio for making this happen! The code is open-source and readily available.
www.nature.com/articles/s41...
www.nature.com/articles/s41...

Deep generative AI models analyzing circulating orphan non-coding RNAs enable detection of early-stage lung cancer - Nature Communications
A generative AI model, Orion, learns a robust and generalizable pattern of non-small cell lung cancer from cancer-specific circulating non-coding RNAs. Orion enhances the performance of liquid biopsy ...
www.nature.com
November 22, 2024 at 12:44 AM
Today was a great day! Our paper on using an AI platform for cell-free RNA liquid biopsy is finally published in Nature Communications! Huge thanks to our amazing team at @exai.bio for making this happen! The code is open-source and readily available.
www.nature.com/articles/s41...
www.nature.com/articles/s41...