



Josue Baeza
@josuebaeza.com
Analytical biochemist working in pharma | Understanding biochemistry one spectra at a time | #TeamMassSpec #Proteomics #MassSpectrometry #TeamDIA #rstats | Opinions are my own | josuebaeza.com under construction
Reposted by Josue Baeza

New paper with collaborators at Thermo. How do to online buffer exchange on membrane proteins in detergent micelles and nanodiscs. Enjoy!
pubs.acs.org/doi/full/10....
pubs.acs.org/doi/full/10....
Online Buffer Exchange Enables Automated Membrane Protein Analysis by Native Mass Spectrometry
Membrane proteins represent the majority of clinical drug targets and are actively involved in a range of cellular processes. However, the complexity of membrane mimetics for membrane protein solubili...
pubs.acs.org
November 14, 2023 at 8:34 PM
New paper with collaborators at Thermo. How do to online buffer exchange on membrane proteins in detergent micelles and nanodiscs. Enjoy!
pubs.acs.org/doi/full/10....
pubs.acs.org/doi/full/10....
Reposted by Josue Baeza

Do you or your lab use the Skyline software for #MassSpectrometry #proteomics? I'm looking for instructors to help with this year's Skyline Online, a virtual workshop/crash-course for all things Skyline! I'm especially looking for early career researchers for this opportunity. Please DM!
September 6, 2025 at 5:11 PM
Do you or your lab use the Skyline software for #MassSpectrometry #proteomics? I'm looking for instructors to help with this year's Skyline Online, a virtual workshop/crash-course for all things Skyline! I'm especially looking for early career researchers for this opportunity. Please DM!
Reposted by Josue Baeza

With the #ASMS2025 app now available, here is a quick summary of what I've found so far from the abstracts:
Sciex - New ZenoTOF 8600, Echo-DMS
Thermo - New Exploris, New Astral (one of these is named Excedion), New OptiFlow source
Agilent - New single quad
Bruker - OmniTIMS, new triple quad, new LC
Sciex - New ZenoTOF 8600, Echo-DMS
Thermo - New Exploris, New Astral (one of these is named Excedion), New OptiFlow source
Agilent - New single quad
Bruker - OmniTIMS, new triple quad, new LC
April 21, 2025 at 8:06 PM
With the #ASMS2025 app now available, here is a quick summary of what I've found so far from the abstracts:
Sciex - New ZenoTOF 8600, Echo-DMS
Thermo - New Exploris, New Astral (one of these is named Excedion), New OptiFlow source
Agilent - New single quad
Bruker - OmniTIMS, new triple quad, new LC
Sciex - New ZenoTOF 8600, Echo-DMS
Thermo - New Exploris, New Astral (one of these is named Excedion), New OptiFlow source
Agilent - New single quad
Bruker - OmniTIMS, new triple quad, new LC
Reposted by Josue Baeza

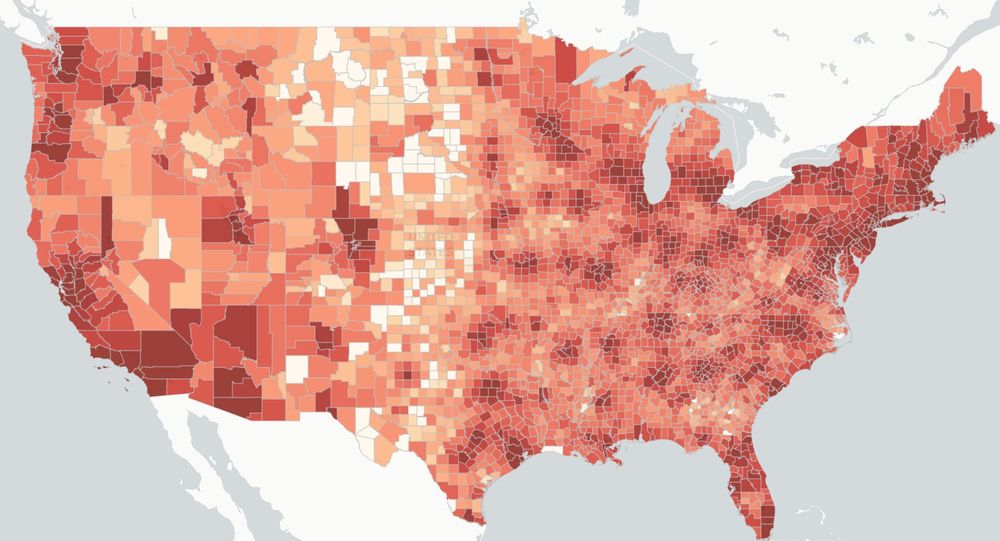
Working with an interdisciplinary team, we have developed a website to communicate how the White House's proposed cuts to health research would cause losses of $16B and 68,500 jobs.
Find out how your community may be impacted.
Explore more at SCIMaP: scienceimpacts.org
a 🧵
Find out how your community may be impacted.
Explore more at SCIMaP: scienceimpacts.org
a 🧵
March 28, 2025 at 2:15 AM
Working with an interdisciplinary team, we have developed a website to communicate how the White House's proposed cuts to health research would cause losses of $16B and 68,500 jobs.
Find out how your community may be impacted.
Explore more at SCIMaP: scienceimpacts.org
a 🧵
Find out how your community may be impacted.
Explore more at SCIMaP: scienceimpacts.org
a 🧵
Reposted by Josue Baeza

I got your waste, fraud, and abuse right here

March 8, 2025 at 2:26 PM
I got your waste, fraud, and abuse right here
Reposted by Josue Baeza

One of the most powerful moments of the day came from Emily Whitehead who told the story of how she was the first pediatric patient to receive CAR T-cell therapy for her leukemia at age 5: “I stand up for science because science saved my life. And that’s a fact.” @standupforscience.bsky.social

March 8, 2025 at 1:18 PM
One of the most powerful moments of the day came from Emily Whitehead who told the story of how she was the first pediatric patient to receive CAR T-cell therapy for her leukemia at age 5: “I stand up for science because science saved my life. And that’s a fact.” @standupforscience.bsky.social
Reposted by Josue Baeza

Join the #StandUpForScience March because science is for everyone.

March 3, 2025 at 10:00 PM
Join the #StandUpForScience March because science is for everyone.
Reposted by Josue Baeza

I have been trying to find the time to move away from the polical hellscape we find ourselves in to finish and share a bluetorial about science.
This helps me remember what this is all about.
Ironically, it is about the treatment of pain.
This helps me remember what this is all about.
Ironically, it is about the treatment of pain.

the word irony is written on a white background
ALT: the word irony is written on a white background
media.tenor.com
February 22, 2025 at 10:19 AM
I have been trying to find the time to move away from the polical hellscape we find ourselves in to finish and share a bluetorial about science.
This helps me remember what this is all about.
Ironically, it is about the treatment of pain.
This helps me remember what this is all about.
Ironically, it is about the treatment of pain.
Reposted by Josue Baeza

News in Proteomics Research blog post | Holy crap - US HUPO starts this week (Saturday! 2/22/25)! proteomicsnews.blogs...
---
#proteomics #prot-other
---
#proteomics #prot-other

February 16, 2025 at 4:40 PM
News in Proteomics Research blog post | Holy crap - US HUPO starts this week (Saturday! 2/22/25)! proteomicsnews.blogs...
---
#proteomics #prot-other
---
#proteomics #prot-other
Reposted by Josue Baeza

Weather’s going wild, and now your mass spec data’s a mess too?
Coindidence? Nope!
We reveal how weather-driven air pressure fluctuations impact diaPASEF-based high troughput proteomics - and how to fix it!
Check out our new paper! #diaPASEF #Weatheromics
pubs.acs.org/doi/10.1021/...

Impact of Local Air Pressure on Ion Mobilities and Data Consistency in diaPASEF-Based High Throughput Proteomics
Data-independent acquisition (DIA) on ion mobility mass spectrometers enables deep proteome coverage and high data completeness in large-scale proteomics studies. For advanced acquisition schemes such as parallel accumulation serial fragmentation-based DIA (diaPASEF) stability of ion mobility (1/K0) over time is crucial for consistent data quality. We found that minor changes in environmental air pressure systematically affect the vacuum pressure in the TIMS analyzer, causing ion mobility shifts. By comparing experimental ion mobilities with historical weather data, we attributed observed drifts to fluctuations in the ground air pressure. Moderate air pressure changes of e.g. fifteen mbar induce ion mobility shifts of 0.025 Vs/cm2. These drifts negatively impact peptide quantification across consecutively acquired samples due to drift-dependent abundance changes and increased missing values for ions located at the boundaries of diaPASEF isolation windows, which cannot be corrected by postprocessing. To address this, we applied an in-batch mobility autocalibration feature on a run-wise basis, leading to full elimination of ion mobility drifts.
pubs.acs.org
January 24, 2025 at 3:35 PM
Weather’s going wild, and now your mass spec data’s a mess too?
Coindidence? Nope!
We reveal how weather-driven air pressure fluctuations impact diaPASEF-based high troughput proteomics - and how to fix it!
Check out our new paper! #diaPASEF #Weatheromics
pubs.acs.org/doi/10.1021/...
Reposted by Josue Baeza

Mass spec continues its relentless world takeover:

Real High Throughput Chemistry Through Mass Spec
www.science.org
December 13, 2024 at 7:36 PM
Mass spec continues its relentless world takeover:
Reposted by Josue Baeza

In the early 2000s, Don Hunt's lab at UVa started developing mass spec based approaches to characterize histone PTMs. I and others were fortunate to be at the right place at the right time back then. Read an account about those early days here.
www.mcponline.org/article/S153...
www.mcponline.org/article/S153...
On the Hunt for the Histone Code
In BriefIn the early 2000s before high resolution mass spectrometers were readily available, the Hunt lab developed many approaches to analyze histone modifications on low resolution instruments.
www.mcponline.org
December 12, 2024 at 2:30 PM
In the early 2000s, Don Hunt's lab at UVa started developing mass spec based approaches to characterize histone PTMs. I and others were fortunate to be at the right place at the right time back then. Read an account about those early days here.
www.mcponline.org/article/S153...
www.mcponline.org/article/S153...
Reposted by Josue Baeza

Quite an exciting read.. everything we're doing today , from instrumentation to analytical methods (2DGE/ IMAC) to recent developments and interest in immunopeptidomics..all had roots in one place. An origins story told very well!
www.mcponline.org/article/S153...
www.mcponline.org/article/S153...
A Donald F. Hunt Story (John’s Version)
In BriefA personal narrative is provided on Donald Hunt’s development of tandem mass spectrometry methods for sequencing peptides.
www.mcponline.org
December 10, 2024 at 6:59 AM
Quite an exciting read.. everything we're doing today , from instrumentation to analytical methods (2DGE/ IMAC) to recent developments and interest in immunopeptidomics..all had roots in one place. An origins story told very well!
www.mcponline.org/article/S153...
www.mcponline.org/article/S153...
Reposted by Josue Baeza

Recently, We saw a discussion on the role of open-source in proteomics. Here, experienced developers & researchers maintaining OS tools for years shared this comment to guide newcomers in the field about OS and its role in the field. 💻 #Proteomics #OpenSource chemrxiv.org/engage/chemr...

Open-source and FAIR Research Software for Proteomics
Scientific discovery relies on innovative software as much as experimental methods, especially in proteomics, where computational tools are essential for mass spectrometer setup, data analysis, and in...
chemrxiv.org
December 9, 2024 at 1:03 PM
Recently, We saw a discussion on the role of open-source in proteomics. Here, experienced developers & researchers maintaining OS tools for years shared this comment to guide newcomers in the field about OS and its role in the field. 💻 #Proteomics #OpenSource chemrxiv.org/engage/chemr...
Reposted by Josue Baeza
Tomorrow is the last day to apply! If you're seeking family and child care support for the annual conference, don’t forget to apply for the US HUPO 2025 Family and Child Care Support Award. Visit ow.ly/m7uE50U5IAu for more details. #USHUPO2025

December 5, 2024 at 5:00 PM
Tomorrow is the last day to apply! If you're seeking family and child care support for the annual conference, don’t forget to apply for the US HUPO 2025 Family and Child Care Support Award. Visit ow.ly/m7uE50U5IAu for more details. #USHUPO2025
Reposted by Josue Baeza

Re-posting our new preprint on match between runs. This multi-lab effort (Keich, Noble, Payne & Smith) led by Alex Solivais should be of interest to anyone doing LFQ. We describe here how to control FDR in LFQ and provide the open source software to do it.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...

Improved detection of differentially abundant proteins through FDR-control of peptide-identity-propagation
Quantitative analysis of proteomics data frequently employs peptide-identity-propagation (PIP) — also known as match-between-runs (MBR) — to increase the number of peptides quantified in a given LC-MS/MS experiment. PIP can routinely account for up to 40% of all quantitative results, with that proportion rising as high as 75% in single-cell proteomics. Therefore, a significant concern for any PIP method is the possibility of false discoveries: errors that result in peptides being quantified incorrectly. Although several tools for label-free quantification (LFQ) claim to control the false discovery rate (FDR) of PIP, these claims cannot be validated as there is currently no accepted method to assess the accuracy of the stated FDR. We present a method for FDR control of PIP, called “PIP-ECHO” (PIP Error Control via Hybrid cOmpetition) and devise a rigorous protocol for evaluating FDR control of any PIP method. Using three different datasets, we evaluate PIP-ECHO alongside the PIP procedures implemented by FlashLFQ, IonQuant, and MaxQuant. These analyses show that PIP-ECHO can accurately control the FDR of PIP at 1% across multiple datasets. Only PIP-ECHO was able to control the FDR in data with injected sample size equivalent to a single-cell dataset. The three other methods fail to control the FDR at 1%, yielding false discovery proportions ranging from 2–6%. We demonstrate the practical implications of this work by performing differential expression analyses on spike-in datasets, where different known amounts of yeast or E. coli peptides are added to a constant background of HeLa cell lysate peptides. In this setting, PIP-ECHO increases both the accuracy and sensitivity of differential expression analysis: our implementation of PIP-ECHO within FlashLFQ enables the detection of 53% more differentially abundant proteins than MaxQuant and 146% more than IonQuant in the spike-in dataset. ### Competing Interest Statement The authors have declared no competing interest.
www.biorxiv.org
December 2, 2024 at 5:05 PM
Re-posting our new preprint on match between runs. This multi-lab effort (Keich, Noble, Payne & Smith) led by Alex Solivais should be of interest to anyone doing LFQ. We describe here how to control FDR in LFQ and provide the open source software to do it.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
Reposted by Josue Baeza

Question for my proteomics skywalkers: today I was chatting about enzymes that aren’t trypsin, and I know there’s plenty out there (including this fun paper, h/t @pastelbio.bsky.social), but any great ones that should be on my radar for bottom-up proteomics?
pubs.acs.org/doi/10.1021/...
pubs.acs.org/doi/10.1021/...

Postproline Cleaving Enzymes also Show Specificity to Reduced Cysteine
In proteomics, postproline cleaving enzymes (PPCEs), such as Aspergillus niger prolyl endopeptidase (AnPEP) and neprosin, complement proteolytic tools because proline is a stop site for many proteases...
pubs.acs.org
November 20, 2024 at 4:41 AM
Question for my proteomics skywalkers: today I was chatting about enzymes that aren’t trypsin, and I know there’s plenty out there (including this fun paper, h/t @pastelbio.bsky.social), but any great ones that should be on my radar for bottom-up proteomics?
pubs.acs.org/doi/10.1021/...
pubs.acs.org/doi/10.1021/...
Reposted by Josue Baeza
Here is the back story behind our recent de novo sequencing benchmark. Science involves a lot of trial and error!
communities.springernature.com/posts/wrangl...
communities.springernature.com/posts/wrangl...
Wrangling a de novo sequencing benchmark
In any machine learning study, high quality data for training and validating the model is critical. This paper describes the result of an iterative process of data wrangling and quality control, which...
communities.springernature.com
November 12, 2024 at 5:00 AM
Here is the back story behind our recent de novo sequencing benchmark. Science involves a lot of trial and error!
communities.springernature.com/posts/wrangl...
communities.springernature.com/posts/wrangl...
Reposted by Josue Baeza

www.nature.com/articles/s41...
2.8 million high-confidence peptide-spectrum matches derived from nine different species from wnoble.bsky.social for #teammassspec
2.8 million high-confidence peptide-spectrum matches derived from nine different species from wnoble.bsky.social for #teammassspec

A multi-species benchmark for training and validating mass spectrometry proteomics machine learning models - Scientific Data
Scientific Data - A multi-species benchmark for training and validating mass spectrometry proteomics machine learning models
www.nature.com
November 17, 2024 at 7:32 AM
www.nature.com/articles/s41...
2.8 million high-confidence peptide-spectrum matches derived from nine different species from wnoble.bsky.social for #teammassspec
2.8 million high-confidence peptide-spectrum matches derived from nine different species from wnoble.bsky.social for #teammassspec
To all the #TeamMassSpec, when using aluminum foil for cooking/grilling/smoking, do you instinctively use the dull side up, or are you normal?
November 21, 2023 at 8:50 PM
To all the #TeamMassSpec, when using aluminum foil for cooking/grilling/smoking, do you instinctively use the dull side up, or are you normal?