


wnoble.bsky.social
@wnoble.bsky.social
As far as I can tell, the main point of this new NIH policy is that there will no longer be an explicit payline in any of the institutes. It's hard to see how this new policy makes the decision-making process "clearer for applicants."
grants.nih.gov/news-events/...
grants.nih.gov/news-events/...
Implementing a Unified NIH Funding Strategy to Guide Consistent and Clearer Award Decisions | Grants & Funding
grants.nih.gov
December 11, 2025 at 7:08 AM
As far as I can tell, the main point of this new NIH policy is that there will no longer be an explicit payline in any of the institutes. It's hard to see how this new policy makes the decision-making process "clearer for applicants."
grants.nih.gov/news-events/...
grants.nih.gov/news-events/...
Reposted by wnoble.bsky.social

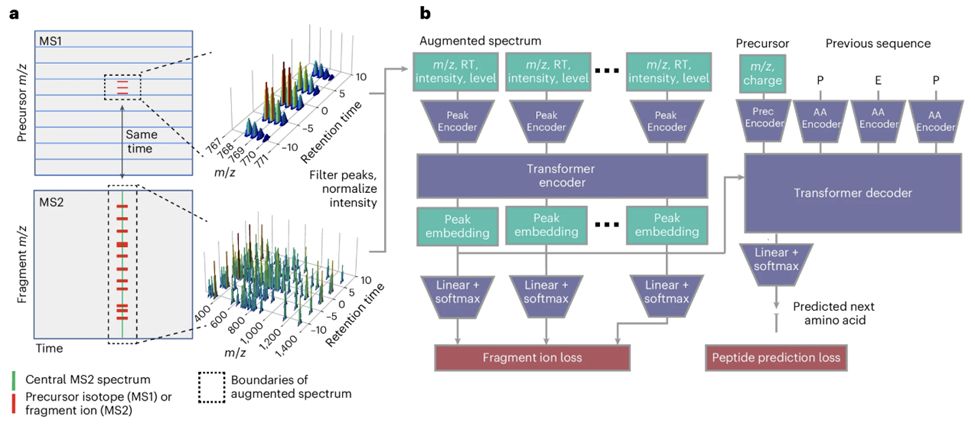
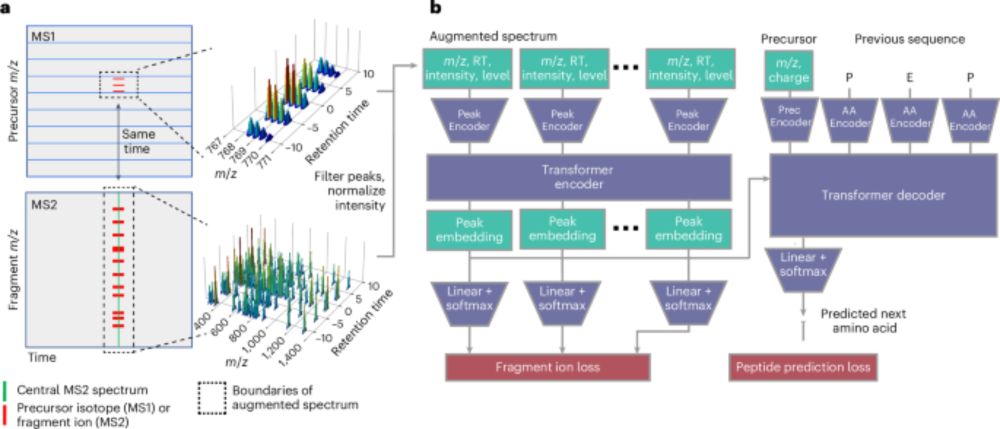
Cascadia from @wnoble.bsky.social is a mass spec-based de novo sequencing model that uses a transformer architecture to handle data-independent acquisition data and achieves substantially improved performance across a range of instruments and experimental protocols. www.nature.com/articles/s41...
July 7, 2025 at 10:31 PM
Cascadia from @wnoble.bsky.social is a mass spec-based de novo sequencing model that uses a transformer architecture to handle data-independent acquisition data and achieves substantially improved performance across a range of instruments and experimental protocols. www.nature.com/articles/s41...
We’re excited to announce the publication of Cascadia, our new de novo sequencing model designed for DIA data. By extending the transformer architecture to fully capture the complexities of DIA data, we achieve state-of-the-art performance.
www.nature.com/articles/s41...
www.nature.com/articles/s41...
A transformer model for de novo sequencing of data-independent acquisition mass spectrometry data - Nature Methods
Cascadia is a mass spectrometry-based de novo sequencing model that uses a transformer architecture to handle data-independent acquisition data and achieves substantially improved performance across a...
www.nature.com
July 3, 2025 at 1:34 AM
We’re excited to announce the publication of Cascadia, our new de novo sequencing model designed for DIA data. By extending the transformer architecture to fully capture the complexities of DIA data, we achieve state-of-the-art performance.
www.nature.com/articles/s41...
www.nature.com/articles/s41...
Reposted by wnoble.bsky.social

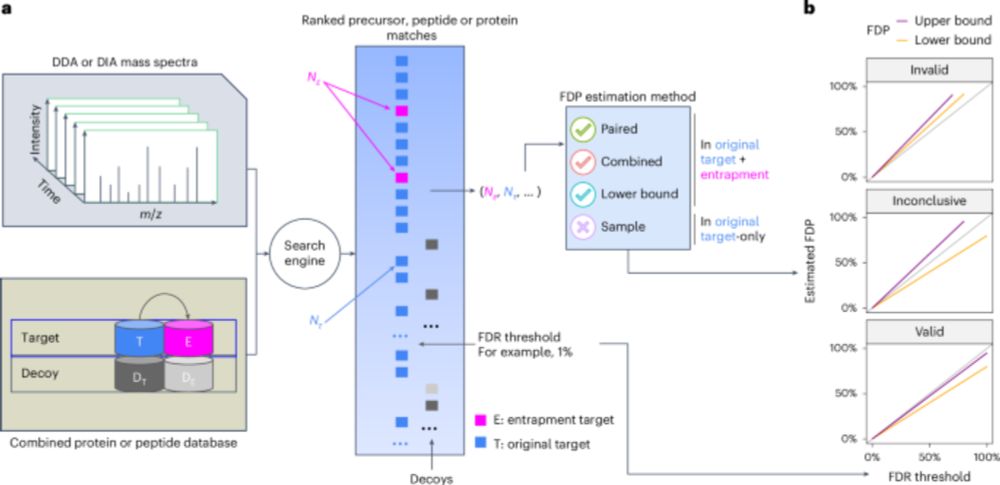
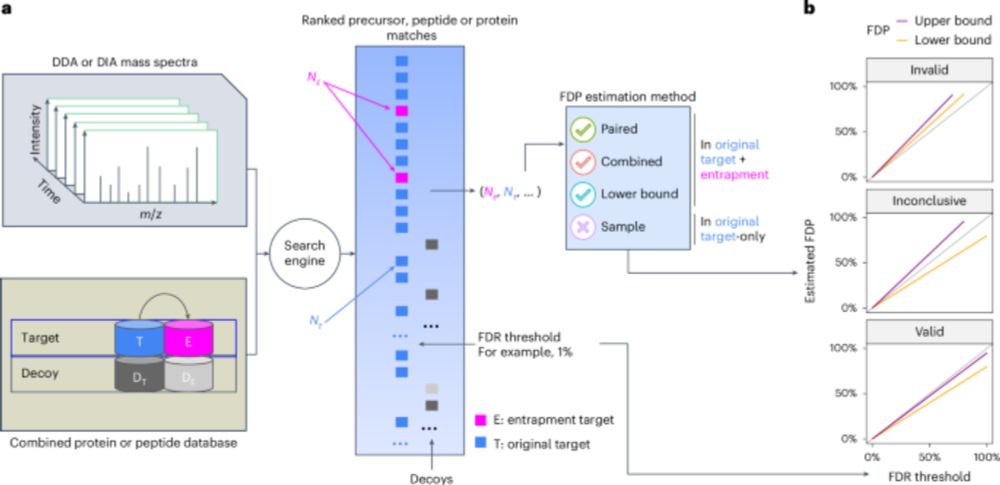
Excited to see this published! It is a good step in the process for people to assess their FDR control in proteomics experiments. Great work from @bo-wen.bsky.social and @urikeich.bsky.social in particular who drove this.
Assessing error control is fundamental in mass spectrometry-based proteomics. @bo-wen.bsky.social @maccoss.bsky.social @urikeich.bsky.social et al introduce a theoretical foundation for entrapment along with a method for more accurate evaluation of FDR control.
www.nature.com/articles/s41...
www.nature.com/articles/s41...
Assessment of false discovery rate control in tandem mass spectrometry analysis using entrapment - Nature Methods
A theoretical foundation for entrapment methods is presented, along with a method that enables more accurate evaluation of false discovery rate (FDR) control in proteomics mass spectrometry analysis p...
www.nature.com
June 16, 2025 at 6:52 PM
Excited to see this published! It is a good step in the process for people to assess their FDR control in proteomics experiments. Great work from @bo-wen.bsky.social and @urikeich.bsky.social in particular who drove this.
Error control in proteomics mass spectrometry analysis is hard. We came up with a way to evaluate error control. Upshot: for old-school DDA data, not so bad. For DIA data, no existing tool successfully controls the false discovery rate!
www.nature.com/articles/s41...
www.nature.com/articles/s41...
Assessment of false discovery rate control in tandem mass spectrometry analysis using entrapment - Nature Methods
A theoretical foundation for entrapment methods is presented, along with a method that enables more accurate evaluation of false discovery rate (FDR) control in proteomics mass spectrometry analysis p...
www.nature.com
June 16, 2025 at 8:57 PM
Error control in proteomics mass spectrometry analysis is hard. We came up with a way to evaluate error control. Upshot: for old-school DDA data, not so bad. For DIA data, no existing tool successfully controls the false discovery rate!
www.nature.com/articles/s41...
www.nature.com/articles/s41...
Interested in prediction tasks involving peptide mass spectra? Our foundation model uses pre-trained spectrum representations learned by a de novo sequencing model to solve many tasks better and with less data, from recognizing chimeras to separating N- and O-glycopeptides. arxiv.org/abs/2505.10848

Foundation model for mass spectrometry proteomics
Mass spectrometry is the dominant technology in the field of proteomics, enabling high-throughput analysis of the protein content of complex biological samples. Due to the complexity of the instrument...
arxiv.org
May 22, 2025 at 11:49 AM
Interested in prediction tasks involving peptide mass spectra? Our foundation model uses pre-trained spectrum representations learned by a de novo sequencing model to solve many tasks better and with less data, from recognizing chimeras to separating N- and O-glycopeptides. arxiv.org/abs/2505.10848
Reposted by wnoble.bsky.social

Ledidi turns any genomics ML model into a controllable sequence designer by inverting the normal ML paradigm. Now, it is significantly faster, flexible, and more powerful than before.
Available on GitHub and installable with `pip install ledidi`
Available on GitHub and installable with `pip install ledidi`
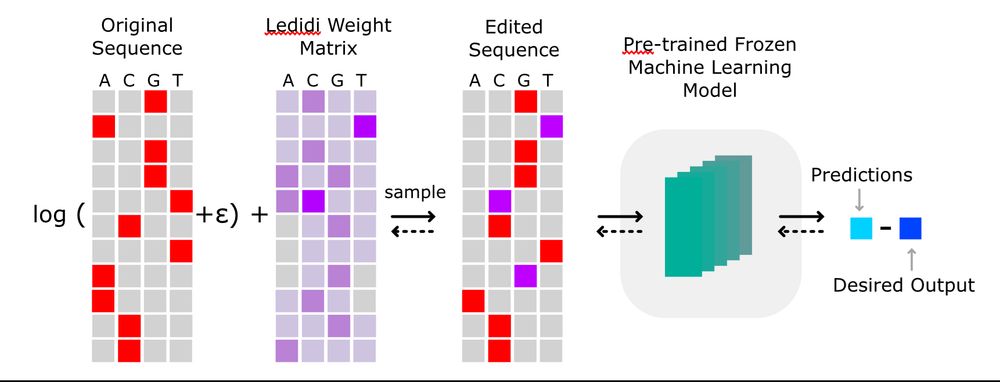
January 7, 2025 at 5:26 PM
Ledidi turns any genomics ML model into a controllable sequence designer by inverting the normal ML paradigm. Now, it is significantly faster, flexible, and more powerful than before.
Available on GitHub and installable with `pip install ledidi`
Available on GitHub and installable with `pip install ledidi`
HiCFoundation is a Swiss army knife for Hi-C data. Any task that takes Hi-C as input will benefit from our pre-trained model. You can do resolution enhancement, reproducibility analysis, loop calling, prediction of epigenomic profiles, or single-cell Hi-C analysis. tinyurl.com/v3nmp6np

A generalizable Hi-C foundation model for chromatin architecture, single-cell and multi-omics analysis across species
Nuclear DNA is organized into a compact three-dimensional (3D) structure that impacts critical cellular processes. High-throughput chromosome conformation capture (Hi-C) is the most widely used method...
tinyurl.com
December 21, 2024 at 12:10 AM
HiCFoundation is a Swiss army knife for Hi-C data. Any task that takes Hi-C as input will benefit from our pre-trained model. You can do resolution enhancement, reproducibility analysis, loop calling, prediction of epigenomic profiles, or single-cell Hi-C analysis. tinyurl.com/v3nmp6np
BLAST is a fantastic tool that has enabled sequence-driven discovery for over 30 years. But, alas, the E-value that it reports turns out to have some serious problems. Here we propose a fix. It's more computationally expensive, but computers are a bit faster than they were in 1990.
bit.ly/3ZDgYt8
bit.ly/3ZDgYt8

A BLAST from the past: revisiting blastp’s E-value
AbstractMotivation. The Basic Local Alignment Search Tool, BLAST, is an indispensable tool for genomic research. BLAST established itself as the canonical
academic.oup.com
December 6, 2024 at 7:24 PM
BLAST is a fantastic tool that has enabled sequence-driven discovery for over 30 years. But, alas, the E-value that it reports turns out to have some serious problems. Here we propose a fix. It's more computationally expensive, but computers are a bit faster than they were in 1990.
bit.ly/3ZDgYt8
bit.ly/3ZDgYt8
Reposted by wnoble.bsky.social

Re-posting our new preprint on match between runs. This multi-lab effort (Keich, Noble, Payne & Smith) led by Alex Solivais should be of interest to anyone doing LFQ. We describe here how to control FDR in LFQ and provide the open source software to do it.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...

Improved detection of differentially abundant proteins through FDR-control of peptide-identity-propagation
Quantitative analysis of proteomics data frequently employs peptide-identity-propagation (PIP) — also known as match-between-runs (MBR) — to increase the number of peptides quantified in a given LC-MS/MS experiment. PIP can routinely account for up to 40% of all quantitative results, with that proportion rising as high as 75% in single-cell proteomics. Therefore, a significant concern for any PIP method is the possibility of false discoveries: errors that result in peptides being quantified incorrectly. Although several tools for label-free quantification (LFQ) claim to control the false discovery rate (FDR) of PIP, these claims cannot be validated as there is currently no accepted method to assess the accuracy of the stated FDR. We present a method for FDR control of PIP, called “PIP-ECHO” (PIP Error Control via Hybrid cOmpetition) and devise a rigorous protocol for evaluating FDR control of any PIP method. Using three different datasets, we evaluate PIP-ECHO alongside the PIP procedures implemented by FlashLFQ, IonQuant, and MaxQuant. These analyses show that PIP-ECHO can accurately control the FDR of PIP at 1% across multiple datasets. Only PIP-ECHO was able to control the FDR in data with injected sample size equivalent to a single-cell dataset. The three other methods fail to control the FDR at 1%, yielding false discovery proportions ranging from 2–6%. We demonstrate the practical implications of this work by performing differential expression analyses on spike-in datasets, where different known amounts of yeast or E. coli peptides are added to a constant background of HeLa cell lysate peptides. In this setting, PIP-ECHO increases both the accuracy and sensitivity of differential expression analysis: our implementation of PIP-ECHO within FlashLFQ enables the detection of 53% more differentially abundant proteins than MaxQuant and 146% more than IonQuant in the spike-in dataset. ### Competing Interest Statement The authors have declared no competing interest.
www.biorxiv.org
December 2, 2024 at 5:05 PM
Re-posting our new preprint on match between runs. This multi-lab effort (Keich, Noble, Payne & Smith) led by Alex Solivais should be of interest to anyone doing LFQ. We describe here how to control FDR in LFQ and provide the open source software to do it.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
How can you transfer peptide IDs between runs and still control your false discovery rate? Till now, the short answer is, you couldn't. Now you can, with PIP-ECHO.
www.biorxiv.org/content/10.1...

Improved detection of differentially abundant proteins through FDR-control of peptide-identity-propagation
Quantitative analysis of proteomics data frequently employs peptide-identity-propagation (PIP) — also known as match-between-runs (MBR) — to increase the number of peptides quantified in a given LC-MS...
www.biorxiv.org
December 2, 2024 at 5:10 PM
How can you transfer peptide IDs between runs and still control your false discovery rate? Till now, the short answer is, you couldn't. Now you can, with PIP-ECHO.
www.biorxiv.org/content/10.1...
Here is the back story behind our recent de novo sequencing benchmark. Science involves a lot of trial and error!
communities.springernature.com/posts/wrangl...
communities.springernature.com/posts/wrangl...
Wrangling a de novo sequencing benchmark
In any machine learning study, high quality data for training and validating the model is critical. This paper describes the result of an iterative process of data wrangling and quality control, which...
communities.springernature.com
November 12, 2024 at 5:00 AM
Here is the back story behind our recent de novo sequencing benchmark. Science involves a lot of trial and error!
communities.springernature.com/posts/wrangl...
communities.springernature.com/posts/wrangl...
Lots of people use machine learning to post process mass spectrometry database search results. But why not just use ML as the score function in database search? Turns out it works great! www.biorxiv.org/content/10.1...

A learned score function improves the power of mass spectrometry database search
bioRxiv - the preprint server for biology, operated by Cold Spring Harbor Laboratory, a research and educational institution
www.biorxiv.org
January 30, 2024 at 10:35 PM
Lots of people use machine learning to post process mass spectrometry database search results. But why not just use ML as the score function in database search? Turns out it works great! www.biorxiv.org/content/10.1...
November 1, 2023 at 4:32 PM
People don’t spend enough time looking at the trans contacts in their Hi-C data. There’s gold in them thar hills! www.biorxiv.org/content/10.1...
October 2, 2023 at 8:13 PM
People don’t spend enough time looking at the trans contacts in their Hi-C data. There’s gold in them thar hills! www.biorxiv.org/content/10.1...