




Youngjae Woo
@youngjaewoo.bsky.social
Computational biologist & Human geneticist |> doing drug discovery
Reposted by Youngjae Woo

We are excited to share GPN-Star, a cost-effective, biologically grounded genomic language modeling framework that achieves state-of-the-art performance across a wide range of variant effect prediction tasks relevant to human genetics.
www.biorxiv.org/content/10.1...
(1/n)
www.biorxiv.org/content/10.1...
(1/n)

September 22, 2025 at 5:29 AM
We are excited to share GPN-Star, a cost-effective, biologically grounded genomic language modeling framework that achieves state-of-the-art performance across a wide range of variant effect prediction tasks relevant to human genetics.
www.biorxiv.org/content/10.1...
(1/n)
www.biorxiv.org/content/10.1...
(1/n)
Reposted by Youngjae Woo
SINGER, our ARG inference method, is finally published and freely available online:
doi.org/10.1038/s415...
It was a long journey – 16 months from initial submission to acceptance. Is it just me, or has peer review gotten more arduous lately? 4+ rounds of review isn't so unusual these days...
doi.org/10.1038/s415...
It was a long journey – 16 months from initial submission to acceptance. Is it just me, or has peer review gotten more arduous lately? 4+ rounds of review isn't so unusual these days...

Robust and accurate Bayesian inference of genome-wide genealogies for hundreds of genomes - Nature Genetics
SINGER is a method for creating ancestral recombination graphs to understand the genealogical history of genomes. The method has increased speed, and thus scalability, without sacrificing accuracy.
doi.org
September 11, 2025 at 3:50 AM
SINGER, our ARG inference method, is finally published and freely available online:
doi.org/10.1038/s415...
It was a long journey – 16 months from initial submission to acceptance. Is it just me, or has peer review gotten more arduous lately? 4+ rounds of review isn't so unusual these days...
doi.org/10.1038/s415...
It was a long journey – 16 months from initial submission to acceptance. Is it just me, or has peer review gotten more arduous lately? 4+ rounds of review isn't so unusual these days...
Reposted by Youngjae Woo

After 1.5 years of work in @kauralasoo.bsky.social’s lab, we finally published my preprint! We introduce gpu-coloc, a GPU-accelerated implementation of coloc, show comparability to CLPP and aim to provide practical guidelines. Now accessible on BioRxiv: www.biorxiv.org/content/10.1...

Ultra-fast genetic colocalisation across millions of traits
Colocalisation is a powerful approach to assess if two genetic association signals are likely to share a causal variant. However, association analyses in large biobanks and molecular quantitative trai...
www.biorxiv.org
August 27, 2025 at 12:19 PM
After 1.5 years of work in @kauralasoo.bsky.social’s lab, we finally published my preprint! We introduce gpu-coloc, a GPU-accelerated implementation of coloc, show comparability to CLPP and aim to provide practical guidelines. Now accessible on BioRxiv: www.biorxiv.org/content/10.1...
Reposted by Youngjae Woo

This is how generative A.I. could be used to design far better, more potent mRNAs (linear and circular) for the future for genome editing, engineered T cells, cancer therapy, vaccines
www.science.org/doi/10.1126/...
@science.org
www.science.org/doi/10.1126/...
@science.org

Deep generative models design mRNA sequences with enhanced translational capacity and stability
Despite the success of mRNA COVID-19 vaccines, extending this modality to more diseases necessitates substantial enhancements. We present GEMORNA, a generative RNA model that utilizes Transformer arch...
www.science.org
August 28, 2025 at 6:28 PM
This is how generative A.I. could be used to design far better, more potent mRNAs (linear and circular) for the future for genome editing, engineered T cells, cancer therapy, vaccines
www.science.org/doi/10.1126/...
@science.org
www.science.org/doi/10.1126/...
@science.org
What predicts your future health better: your DNA or your medical records?
> EHR-based risk scores often outperform PGS for several common diseases and add predictive value even when combined with PGS.
> PGS better in predicting cancer.
www.nature.com/articles/s41...
> EHR-based risk scores often outperform PGS for several common diseases and add predictive value even when combined with PGS.
> PGS better in predicting cancer.
www.nature.com/articles/s41...

Cross-biobank generalizability and accuracy of electronic health record-based predictors compared to polygenic scores - Nature Genetics
Comparison of electronic health record-based phenotype risk scores (PheRS) and polygenic scores (PGS) across 13 common diseases and three biobank-based studies indicates that PheRS and PGS may provide...
www.nature.com
August 27, 2025 at 8:31 PM
What predicts your future health better: your DNA or your medical records?
> EHR-based risk scores often outperform PGS for several common diseases and add predictive value even when combined with PGS.
> PGS better in predicting cancer.
www.nature.com/articles/s41...
> EHR-based risk scores often outperform PGS for several common diseases and add predictive value even when combined with PGS.
> PGS better in predicting cancer.
www.nature.com/articles/s41...
Reposted by Youngjae Woo

Shiny new probabilistic model, gruyere 🧀, for powering up rare variant associations w/ DL effect prediction! We find novel associations for Alzheimer's disease, e.g. nuclear pore protein NUP93 in microglia. Big thanks to NIH/NIA/ADSP and Anjali for the hard work! authors.elsevier.com/a/1ldzwgeXDzHj
authors.elsevier.com
August 20, 2025 at 10:06 PM
Shiny new probabilistic model, gruyere 🧀, for powering up rare variant associations w/ DL effect prediction! We find novel associations for Alzheimer's disease, e.g. nuclear pore protein NUP93 in microglia. Big thanks to NIH/NIA/ADSP and Anjali for the hard work! authors.elsevier.com/a/1ldzwgeXDzHj
The study used plasma proteomics data from 46,665 individuals in the UK Biobank to build large-language model (LLM) predictors for identifying damaging genetic variants, increasing the proportion of missense variants scored with pLoF-like effects.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...

Variant Classification Using Proteomics-Informed Large Language Models Increases Power of Rare Variant Association Studies and Enhances Target Discovery
Rare variant association analysis, which assesses the aggregate effect of rare damaging variants within a gene, is a powerful strategy for advancing knowledge of human biology. Numerous models have be...
www.biorxiv.org
August 8, 2025 at 2:59 PM
The study used plasma proteomics data from 46,665 individuals in the UK Biobank to build large-language model (LLM) predictors for identifying damaging genetic variants, increasing the proportion of missense variants scored with pLoF-like effects.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...

Reposted by Youngjae Woo

We are proud to be a founding contributor to lipidinteractome.org, a repository developed by @tafesselab.bsky.social & Schultz lab to increase accessibility to proteomics data from multi-functionalized lipid analogs! Check out the website & preprint: arxiv.org/abs/2507.23101 #lipidtime
The Lipid Interactome Repository – Lipid Interactome Repository
lipidinteractome.org
August 5, 2025 at 1:06 PM
We are proud to be a founding contributor to lipidinteractome.org, a repository developed by @tafesselab.bsky.social & Schultz lab to increase accessibility to proteomics data from multi-functionalized lipid analogs! Check out the website & preprint: arxiv.org/abs/2507.23101 #lipidtime
Reposted by Youngjae Woo

Excited to share work with
Zhidian Zhang, @milot.bsky.social, @martinsteinegger.bsky.social, and @sokrypton.org
biorxiv.org/content/10.1...
TLDR: We introduce MSA Pairformer, a 111M parameter protein language model that challenges the scaling paradigm in self-supervised protein language modeling🧵
Zhidian Zhang, @milot.bsky.social, @martinsteinegger.bsky.social, and @sokrypton.org
biorxiv.org/content/10.1...
TLDR: We introduce MSA Pairformer, a 111M parameter protein language model that challenges the scaling paradigm in self-supervised protein language modeling🧵

Scaling down protein language modeling with MSA Pairformer
Recent efforts in protein language modeling have focused on scaling single-sequence models and their training data, requiring vast compute resources that limit accessibility. Although models that use ...
biorxiv.org
August 5, 2025 at 6:31 AM
Excited to share work with
Zhidian Zhang, @milot.bsky.social, @martinsteinegger.bsky.social, and @sokrypton.org
biorxiv.org/content/10.1...
TLDR: We introduce MSA Pairformer, a 111M parameter protein language model that challenges the scaling paradigm in self-supervised protein language modeling🧵
Zhidian Zhang, @milot.bsky.social, @martinsteinegger.bsky.social, and @sokrypton.org
biorxiv.org/content/10.1...
TLDR: We introduce MSA Pairformer, a 111M parameter protein language model that challenges the scaling paradigm in self-supervised protein language modeling🧵
Reposted by Youngjae Woo

A few thoughts on Herasight, the new embryo selection company. First, their whitepaper (drive.google.com/file/d/1EpFi...) implies that competitors like Nucleus have been marketing and selling grossly erroneous risk estimates. This is shocking if true! 🧵
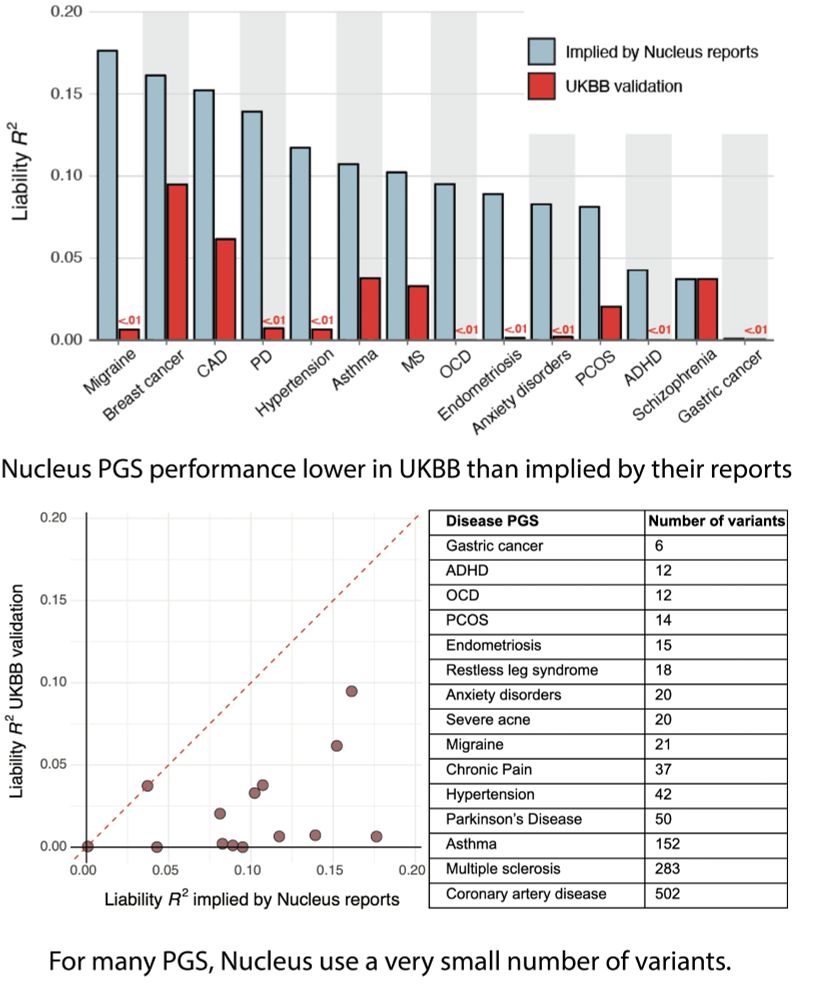
August 2, 2025 at 2:38 PM
A few thoughts on Herasight, the new embryo selection company. First, their whitepaper (drive.google.com/file/d/1EpFi...) implies that competitors like Nucleus have been marketing and selling grossly erroneous risk estimates. This is shocking if true! 🧵
Reposted by Youngjae Woo

Realizing the promise of genome-wide association studies for effector gene prediction www.nature.com/articles/s41...


June 12, 2025 at 10:25 AM
Realizing the promise of genome-wide association studies for effector gene prediction www.nature.com/articles/s41...
Reposted by Youngjae Woo

New preprint in collaboration with @paulinanunezv.bsky.social supervised by @jonnyfrazer.bsky.social and Mafalda Dias – we propose a simple approach to improving zero-shot variant effect prediction in pre-existing protein and genome language models: 🧶 1/n
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...

From Likelihood to Fitness: Improving Variant Effect Prediction in Protein and Genome Language Models
Generative models trained on natural sequences are increasingly used to predict the effects of genetic variation, enabling progress in therapeutic design, disease risk prediction, and synthetic biolog...
www.biorxiv.org
May 26, 2025 at 5:30 PM
New preprint in collaboration with @paulinanunezv.bsky.social supervised by @jonnyfrazer.bsky.social and Mafalda Dias – we propose a simple approach to improving zero-shot variant effect prediction in pre-existing protein and genome language models: 🧶 1/n
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
Reposted by Youngjae Woo
The 2026 Probabilistic Modeling in Genomics (ProbGen) meeting will be held at UC Berkeley, March 25-28, 2026. We have an amazing list of keynote speakers and session chairs:
probgen2026.github.io
Please help spread the news.
probgen2026.github.io
Please help spread the news.
Home - ProbGen 2026
Your Site Description
probgen2026.github.io
June 6, 2025 at 5:52 PM
The 2026 Probabilistic Modeling in Genomics (ProbGen) meeting will be held at UC Berkeley, March 25-28, 2026. We have an amazing list of keynote speakers and session chairs:
probgen2026.github.io
Please help spread the news.
probgen2026.github.io
Please help spread the news.
Reposted by Youngjae Woo

To add to your weekend reading, check out this nice report on the Genes & Health cohort from Hye In Kim, David van Heel and many many more:
www.medrxiv.org/content/10.1...
www.medrxiv.org/content/10.1...

Exome sequencing and analysis of 44,028 British South Asians enriched for high autozygosity
Genes and Health (G&H) is a biomedical study of adult British-Pakistani and -Bangladeshi research volunteers enriched for autozygosity. We performed whole exome sequencing in 44,028 G&H participants, ...
www.medrxiv.org
June 7, 2025 at 3:20 PM
To add to your weekend reading, check out this nice report on the Genes & Health cohort from Hye In Kim, David van Heel and many many more:
www.medrxiv.org/content/10.1...
www.medrxiv.org/content/10.1...
Reposted by Youngjae Woo

Also, Charlie's #VariantEffect25 talk is available here www.youtube.com/watch?v=johJ...
June 7, 2025 at 6:30 PM
Also, Charlie's #VariantEffect25 talk is available here www.youtube.com/watch?v=johJ...
Reposted by Youngjae Woo
Wow. "NIH" canceled my co-mentored (with Dave Sulzer) PhD student's F31 funding. His work is on understanding the genetics and neuroscience of language learning disorders. F31 provides no indirect $ to Columbia, just pays his salary. Not that it should matter, but he's an American citizen. W.T.F.
March 11, 2025 at 12:41 PM
Wow. "NIH" canceled my co-mentored (with Dave Sulzer) PhD student's F31 funding. His work is on understanding the genetics and neuroscience of language learning disorders. F31 provides no indirect $ to Columbia, just pays his salary. Not that it should matter, but he's an American citizen. W.T.F.
Reposted by Youngjae Woo

What do GWAS and rare variant burden tests discover, and why?
Do these studies find the most IMPORTANT genes? If not, how DO they rank genes?
Here we present a surprising result: these studies actually test for SPECIFICITY! A 🧵on what this means... (🧪🧬)
www.biorxiv.org/content/10.1...
Do these studies find the most IMPORTANT genes? If not, how DO they rank genes?
Here we present a surprising result: these studies actually test for SPECIFICITY! A 🧵on what this means... (🧪🧬)
www.biorxiv.org/content/10.1...

Specificity, length, and luck: How genes are prioritized by rare and common variant association studies
Standard genome-wide association studies (GWAS) and rare variant burden tests are essential tools for identifying trait-relevant genes. Although these methods are conceptually similar, we show by anal...
www.biorxiv.org
December 17, 2024 at 7:05 AM
What do GWAS and rare variant burden tests discover, and why?
Do these studies find the most IMPORTANT genes? If not, how DO they rank genes?
Here we present a surprising result: these studies actually test for SPECIFICITY! A 🧵on what this means... (🧪🧬)
www.biorxiv.org/content/10.1...
Do these studies find the most IMPORTANT genes? If not, how DO they rank genes?
Here we present a surprising result: these studies actually test for SPECIFICITY! A 🧵on what this means... (🧪🧬)
www.biorxiv.org/content/10.1...
Reposted by Youngjae Woo

🗨️ WANNA TALK TO YOUR CELLS? Try out CellWhisperer – our new multimodal AI that turns single-cell RNA-seq analysis into a conversation. No coding needed, just chat in plain English. Short walkthrough below. Web app & bioRxiv preprint linked in the thread. Let's dive in! (1/9)
October 18, 2024 at 9:32 AM
🗨️ WANNA TALK TO YOUR CELLS? Try out CellWhisperer – our new multimodal AI that turns single-cell RNA-seq analysis into a conversation. No coding needed, just chat in plain English. Short walkthrough below. Web app & bioRxiv preprint linked in the thread. Let's dive in! (1/9)
Reposted by Youngjae Woo

In a new preprint w collaborators Jiaxin Hu and Miaoyan Wang at Univ Wisconsin
www.biorxiv.org/content/10.1...
we develop a method to map QTL that change gene coexpression networks. For each genetic marker in an F2 cross, split the data by genotype & build a coexpression network for each genotype
www.biorxiv.org/content/10.1...
we develop a method to map QTL that change gene coexpression networks. For each genetic marker in an F2 cross, split the data by genotype & build a coexpression network for each genotype

March 31, 2024 at 12:49 PM
In a new preprint w collaborators Jiaxin Hu and Miaoyan Wang at Univ Wisconsin
www.biorxiv.org/content/10.1...
we develop a method to map QTL that change gene coexpression networks. For each genetic marker in an F2 cross, split the data by genotype & build a coexpression network for each genotype
www.biorxiv.org/content/10.1...
we develop a method to map QTL that change gene coexpression networks. For each genetic marker in an F2 cross, split the data by genotype & build a coexpression network for each genotype
Reposted by Youngjae Woo

Nanopore sequencing of 1000 Genomes Project samples to build a comprehensive catalog of human genetic variation https://www.medrxiv.org/content/10.1101/2024.03.05.24303792v1

Nanopore sequencing of 1000 Genomes Project samples to build a comprehensive catalog of human genetic variation https://www.medrxiv.org/content/10.1101/2024.03.05.24303792v1
Less than half of individuals with a suspected Mendelian condition receive a precise molecular diagn
www.medrxiv.org
March 7, 2024 at 8:40 PM
Nanopore sequencing of 1000 Genomes Project samples to build a comprehensive catalog of human genetic variation https://www.medrxiv.org/content/10.1101/2024.03.05.24303792v1
Reposted by Youngjae Woo

A harmonized public resource of deeply sequenced diverse human genomes www.biorxiv.org/content/10.1... gnomad.broadinstitute.org/news/2020-10...

February 28, 2024 at 11:01 AM
A harmonized public resource of deeply sequenced diverse human genomes www.biorxiv.org/content/10.1... gnomad.broadinstitute.org/news/2020-10...
Reposted by Youngjae Woo

Excited to share our review paper in Cell! With @sashagusevposts.bsky.social @sramach.bsky.social and Yang Li we discuss the genetic and molecular architecture of human traits, future opportunities, challenges and ways forward.
authors.elsevier.com/a/1igrbL7PXq...
authors.elsevier.com/a/1igrbL7PXq...
February 29, 2024 at 4:00 PM
Excited to share our review paper in Cell! With @sashagusevposts.bsky.social @sramach.bsky.social and Yang Li we discuss the genetic and molecular architecture of human traits, future opportunities, challenges and ways forward.
authors.elsevier.com/a/1igrbL7PXq...
authors.elsevier.com/a/1igrbL7PXq...
Reposted by Youngjae Woo
Some thoughts on the ability to distinguish populations with genetic variation, why that means little for trait differences, and why there are other good reasons to collect diverse data:
threadreaderapp.com/thread/17633...
threadreaderapp.com/thread/17633...
March 1, 2024 at 1:40 AM
Some thoughts on the ability to distinguish populations with genetic variation, why that means little for trait differences, and why there are other good reasons to collect diverse data:
threadreaderapp.com/thread/17633...
threadreaderapp.com/thread/17633...
Reposted by Youngjae Woo

Causal mediation analysis for time-varying heritable risk factors with Mendelian Randomization https://www.biorxiv.org/content/10.1101/2024.02.10.579129v1

Causal mediation analysis for time-varying heritable risk factors with Mendelian Randomization https://www.biorxiv.org/content/10.1101/2024.02.10.579129v1
Understanding the causal pathogenic mechanisms of diseases is crucial in clinical research. When ran
www.biorxiv.org
February 12, 2024 at 11:47 AM
Causal mediation analysis for time-varying heritable risk factors with Mendelian Randomization https://www.biorxiv.org/content/10.1101/2024.02.10.579129v1