



Rafael Najmanovich
@rnajmanovich.bsky.social
Structural bioinformatics, computational biophysics, drug design, protein dynamics at University of Montreal. In my free time I create bonsai trees and care for these and other fantastic beasts (reptiles, amphibians and tarantulas).
It was a great meeting. Thank you @gonzaparra.bsky.social for making sure every detail worked out - except for the rain... :)
Everything comes to an end but memories remain!!
We had a great 3DSIG/3DBioinfo event in "sunny" Barcelona this week!! I have enjoyed so much to help organising it & to have such great minds all together sharing science for 3 days!!
Aufwiedersehen friends! For more science & friendships to come! ⭐
We had a great 3DSIG/3DBioinfo event in "sunny" Barcelona this week!! I have enjoyed so much to help organising it & to have such great minds all together sharing science for 3 days!!
Aufwiedersehen friends! For more science & friendships to come! ⭐


March 24, 2025 at 12:48 AM
It was a great meeting. Thank you @gonzaparra.bsky.social for making sure every detail worked out - except for the rain... :)
Reposted by Rafael Najmanovich

So on March 9, a French scientist, on a visa to attend a conference in Houston, was denied entry to the US and subsequently expelled because his phone had messages decrying scientific policies put forward by the Trump administration: www.lemonde.fr/internationa...

Etats-Unis : un chercheur français refoulé pour avoir exprimé « une opinion personnelle sur la politique menée par l’administration Trump »
Le ministre de la recherche français a dit sa « préoccupation », mercredi, après cette décision des autorités américaines. Le chercheur du CNRS aurait subi un contrôle aléatoire à son arrivée, avant q...
www.lemonde.fr
March 19, 2025 at 6:31 PM
So on March 9, a French scientist, on a visa to attend a conference in Houston, was denied entry to the US and subsequently expelled because his phone had messages decrying scientific policies put forward by the Trump administration: www.lemonde.fr/internationa...
NRGRank, with which you can screen 10^6 molecules per day in a modern laptop, is out. Particularly useful for apo form or AlphaFold models but in all cases (including hole), finding binders that are missed by Glide (the opposite is also true). NRGRank requires 0.3s/compound - 100-1000 fold faster.

March 9, 2025 at 1:58 AM
NRGRank, with which you can screen 10^6 molecules per day in a modern laptop, is out. Particularly useful for apo form or AlphaFold models but in all cases (including hole), finding binders that are missed by Glide (the opposite is also true). NRGRank requires 0.3s/compound - 100-1000 fold faster.
Happy to share our latest preprint. With NRGRank you can screen 1M in a day in a laptop (use our NEGSuite-Qt for that) - a billion compounds per day with our computational resources.

NRGRank: Coarse-grained structurally-informed ultra-massive virtual screening https://www.biorxiv.org/content/10.1101/2025.02.17.638675v1
February 22, 2025 at 11:26 AM
Happy to share our latest preprint. With NRGRank you can screen 1M in a day in a laptop (use our NEGSuite-Qt for that) - a billion compounds per day with our computational resources.
"We demonstrate that current co-folding approaches largely memorise ligand poses from their training data, hindering their use for de novo drug design."

Excited to share our latest preprint evaluating AlphaFold3, Boltz-1, Chai-1 and Protenix for predicting protein-ligand interactions, featuring our newly introduced benchmark dataset 🌹Runs N’ Poses🌹!
www.biorxiv.org/content/10.1...
🧵👇 (1/n)
www.biorxiv.org/content/10.1...
🧵👇 (1/n)

Have protein-ligand co-folding methods moved beyond memorisation?
Deep learning has driven major breakthroughs in protein structure prediction, however the next critical advance is accurately predicting how proteins interact with other molecules, especially small mo...
www.biorxiv.org
February 10, 2025 at 2:26 PM
"We demonstrate that current co-folding approaches largely memorise ligand poses from their training data, hindering their use for de novo drug design."
Reposted by Rafael Najmanovich

🚨🚨 MEGA JOB ALERT 🚨🚨
Independent Group Leader Positions in Computational Biology @humantechnopole.bsky.social!
Are you ready to start your own lab? Do you know someone who is? Repost this + share with everyone who might want to know about it. Thanks!!! 🙏
More details below... check it out! 🧵 1/3
Independent Group Leader Positions in Computational Biology @humantechnopole.bsky.social!
Are you ready to start your own lab? Do you know someone who is? Repost this + share with everyone who might want to know about it. Thanks!!! 🙏
More details below... check it out! 🧵 1/3

January 29, 2025 at 10:40 AM
🚨🚨 MEGA JOB ALERT 🚨🚨
Independent Group Leader Positions in Computational Biology @humantechnopole.bsky.social!
Are you ready to start your own lab? Do you know someone who is? Repost this + share with everyone who might want to know about it. Thanks!!! 🙏
More details below... check it out! 🧵 1/3
Independent Group Leader Positions in Computational Biology @humantechnopole.bsky.social!
Are you ready to start your own lab? Do you know someone who is? Repost this + share with everyone who might want to know about it. Thanks!!! 🙏
More details below... check it out! 🧵 1/3
Reposted by Rafael Najmanovich

In The Pipeline blog featuring estrogen receptor papers with contributions from @nrimpact.bsky.social members @docfanning.bsky.social and @nelsonlab.bsky.social #nuclearreceptors

This one is mostly about the title subject (featuring a very interesting compound from academia) but it’s also about attacking the NIH:

Attacking Estrogen-Receptor-Positive Breast Cancer
www.science.org
January 24, 2025 at 7:20 PM
In The Pipeline blog featuring estrogen receptor papers with contributions from @nrimpact.bsky.social members @docfanning.bsky.social and @nelsonlab.bsky.social #nuclearreceptors
I am following up on the previous post with a little more detail. The NRGSute-Qt is a PyMOL plugin to use our major computational tools. Tools that offer high-performance, speed and ease of use. (1/N)

NRGSuite-Qt: A PyMOL plugin for high-throughput virtual screening, molecular docking, normal-mode analysis, the study of molecular interactions and the detection of binding-site similarities
We introduce NRGSuite-Qt, a PyMOL plugin that provides a comprehensive toolkit for protein modeling, virtual screening, normal mode analysis, and binding-site similarity calculations. Building on the ...
doi.org
January 24, 2025 at 7:52 PM
I am following up on the previous post with a little more detail. The NRGSute-Qt is a PyMOL plugin to use our major computational tools. Tools that offer high-performance, speed and ease of use. (1/N)
Our newest preprint is out. A PyMOL plugin giving access to a broad suite of high-performance, fast, and easy to use methods for virtual screening (50K molecules/h), docking, analysis of molecular interactions, dynamics, engineering permitting complex workflows.
NRGSuite-Qt: A PyMOL plugin for high-throughput virtual screening, molecular docking, normal-mode analysis, the study of molecular interactions and the detection of binding-site similarities https://www.biorxiv.org/content/10.1101/2025.01.23.634566v1
January 24, 2025 at 2:38 PM
Our newest preprint is out. A PyMOL plugin giving access to a broad suite of high-performance, fast, and easy to use methods for virtual screening (50K molecules/h), docking, analysis of molecular interactions, dynamics, engineering permitting complex workflows.
Anyone else experiencing problems with @biorxivpreprint.bsky.social ? Like preprints with mismatched abstracts? it is happening to my latest: www.biorxiv.org/content/10.1... - although when originally published it was all fine.
Comprehensive Analysis of SARS-CoV-2 Spike Evolution: Epitope Classification and Immune Escape Prediction
The evolution of SARS-CoV-2, the virus responsible for the COVID-19 pandemic, has produced unprece-dented numbers of structures of the Spike protein. This study presents a comprehensive analysis of 1,...
www.biorxiv.org
December 19, 2024 at 2:37 PM
Anyone else experiencing problems with @biorxivpreprint.bsky.social ? Like preprints with mismatched abstracts? it is happening to my latest: www.biorxiv.org/content/10.1... - although when originally published it was all fine.
Reposted by Rafael Najmanovich

After listening to community feedback, we will provide a partial feed allowing 93% of eLife authors to continue being indexed in the Web of Science Core Collection. We will also move from the Scopus Journals Collection to the Scopus Preprints Collection.
Full update below.
Full update below.

Changes to eLife’s indexing status in Web of Science and Scopus
To best serve the needs of researchers, eLife will provide a partial feed of research to be indexed in the Web of Science Core Collection. eLife will also move from the Scopus Journals Collection to…
buff.ly
December 10, 2024 at 3:14 PM
After listening to community feedback, we will provide a partial feed allowing 93% of eLife authors to continue being indexed in the Web of Science Core Collection. We will also move from the Scopus Journals Collection to the Scopus Preprints Collection.
Full update below.
Full update below.
and also our latest work, Natálias latest preprint, a collaboration with the groups of
@mattbashton.bsky.social and Ricardo Rajsbaum is out in BioRxiv. www.biorxiv.org/content/10.1...
@mattbashton.bsky.social and Ricardo Rajsbaum is out in BioRxiv. www.biorxiv.org/content/10.1...
Comprehensive Analysis of SARS-CoV-2 Spike Evolution: Epitope Classification and Immune Escape Prediction
The evolution of SARS-CoV-2, the virus responsible for the COVID-19 pandemic, has produced unprecedented numbers of structures of the Spike protein. This study presents a comprehensive analysis of 1,560 published Spike protein structures, capturing most variants that emerged throughout the pandemic and covering diverse heteromerization and interacting complexes. We employ an interaction-energy informed geometric clustering to identify 14 epitopes characterized by their conformational specificity, shared interface with ACE2 binding, and glycosylation patterns. Our per-residue interaction evaluations accurately predict each residue's role in antibody recognition and as well as experimental measurements of immune escape, showing strong correlations with DMS data, thus making it possible to predict the behaviour of future variants. We integrate the structural analysis with a longitudinal analysis of nearly 3 million viral sequences. This broad-ranging structural and longitudinal analysis provides insight into the effect of specific mutations on the energetics of interactions and dynamics of the SARS-CoV-2 Spike protein during the course of the pandemic. Specifically, with the emergence of widespread immunity, we observe an enthalpic trade-off in which mutations in the receptor binding motif (RBM) that promote immune escape also weaken the interaction with ACE2. Additionally, we also observe a second mechanism, that we call entropic trade-off, in which mutations outside of the RBM contribute to decrease the occupancy of the open state of SARS-CoV-2 Spike, thus also contributing to immune escape at the expense of ACE2 binding but without changes on the ACE2 binding interface. This work not only highlights the role of mutations across SARS-CoV-2 Spike variants but also reveals the complex interplay of evolutionary forces shaping the evolution of the SARS-CoV-2 Spike protein over the course of the pandemic. ### Competing Interest Statement The authors have declared no competing interest.
www.biorxiv.org
December 10, 2024 at 12:02 PM
and also our latest work, Natálias latest preprint, a collaboration with the groups of
@mattbashton.bsky.social and Ricardo Rajsbaum is out in BioRxiv. www.biorxiv.org/content/10.1...
@mattbashton.bsky.social and Ricardo Rajsbaum is out in BioRxiv. www.biorxiv.org/content/10.1...
Congratulations to Dr. Natalia Teruel, who successfully defended her PhD yesterday, her work was judged to be exceptional and worth to be added to the Faculté de médecine - Université de Montréal rector's honours list.

December 10, 2024 at 11:52 AM
Congratulations to Dr. Natalia Teruel, who successfully defended her PhD yesterday, her work was judged to be exceptional and worth to be added to the Faculté de médecine - Université de Montréal rector's honours list.
Fascinating theory of why we dream. time.com/5925206/why-...

Why Do We Dream? A New Theory on How It Protects Our Brains
"Dreams are primarily visual precisely because this is the only sense that is disadvantaged by darkness."
time.com
November 22, 2024 at 2:42 AM
Fascinating theory of why we dream. time.com/5925206/why-...
I am 100% in favour of the eLife publishing model.
Following the news that eLife will not receive an Impact Factor in 2025, we’ve shared an update on how our model is doing since we were first placed “on hold” by Web of Science, and what we’re up to now. Find out more.
https://buff.ly/3ATRAFT
https://buff.ly/3ATRAFT

The eLife Model: An update on progress following changes in Web of Science indexing status
Following the decision that eLife will not receive an Impact Factor in 2025, we share an update on how our model is doing since we were first placed “on hold” by Web of Science, and what we’re up to…
buff.ly
November 21, 2024 at 1:54 AM
I am 100% in favour of the eLife publishing model.
Reposted by Rafael Najmanovich

Big announcement 📢🚨
The ISCB 3DSig together with the 3DBioinfo @ELIXIREurope community we are organising a joint conference in Barcelona March 19-21, 2025!
If you work in Structural Bioinformatics or Computational Biophysics communities register ASAP! Only 120 spots!
www.iscb.org/3dbioinfo202...
The ISCB 3DSig together with the 3DBioinfo @ELIXIREurope community we are organising a joint conference in Barcelona March 19-21, 2025!
If you work in Structural Bioinformatics or Computational Biophysics communities register ASAP! Only 120 spots!
www.iscb.org/3dbioinfo202...

November 19, 2024 at 7:04 PM
Big announcement 📢🚨
The ISCB 3DSig together with the 3DBioinfo @ELIXIREurope community we are organising a joint conference in Barcelona March 19-21, 2025!
If you work in Structural Bioinformatics or Computational Biophysics communities register ASAP! Only 120 spots!
www.iscb.org/3dbioinfo202...
The ISCB 3DSig together with the 3DBioinfo @ELIXIREurope community we are organising a joint conference in Barcelona March 19-21, 2025!
If you work in Structural Bioinformatics or Computational Biophysics communities register ASAP! Only 120 spots!
www.iscb.org/3dbioinfo202...
Reposted by Rafael Najmanovich

From @gonzaparra.bsky.social on the other site: "deeper MSAs allow for better stability predictions as the coevolutionary signal associated with functional motifs gets buffered out & what's left are fold stability anchors" www.nature.com/articles/s41...

Local energetic frustration conservation in protein families and superfamilies - Nature Communications
Energetic local frustration in proteins may have been positively selected by evolution when related to function such as ligand binding, allostery and other. Here the authors present a methodology to a...
www.nature.com
November 10, 2024 at 4:55 PM
From @gonzaparra.bsky.social on the other site: "deeper MSAs allow for better stability predictions as the coevolutionary signal associated with functional motifs gets buffered out & what's left are fold stability anchors" www.nature.com/articles/s41...
Reposted by Rafael Najmanovich

If you find this work interesting I'm currently looking for a new PhD student, see the Add here www.findaphd.com/phds/project...

Trashzymes: Mining Municipal solid waste and alkaliphilic species for brilliant cleaning at Procter & Gamble on FindAPhD.com
PhD Project - Trashzymes: Mining Municipal solid waste and alkaliphilic species for brilliant cleaning at Procter & Gamble, listed on FindAPhD.com
www.findaphd.com
November 14, 2024 at 7:56 PM
If you find this work interesting I'm currently looking for a new PhD student, see the Add here www.findaphd.com/phds/project...
Reposted by Rafael Najmanovich
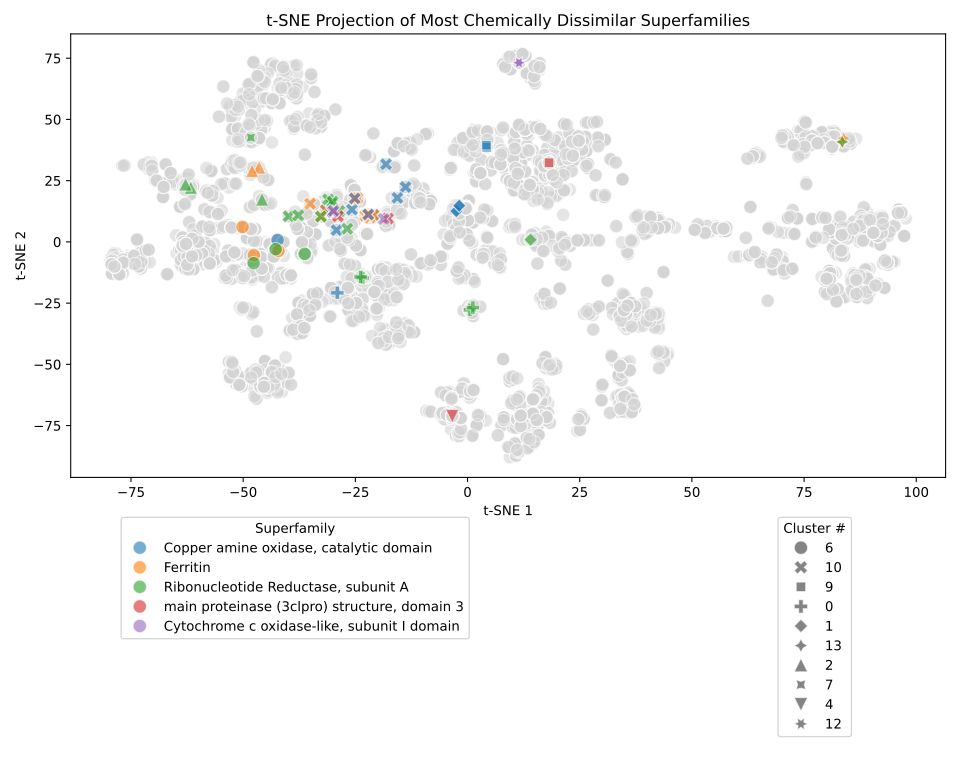
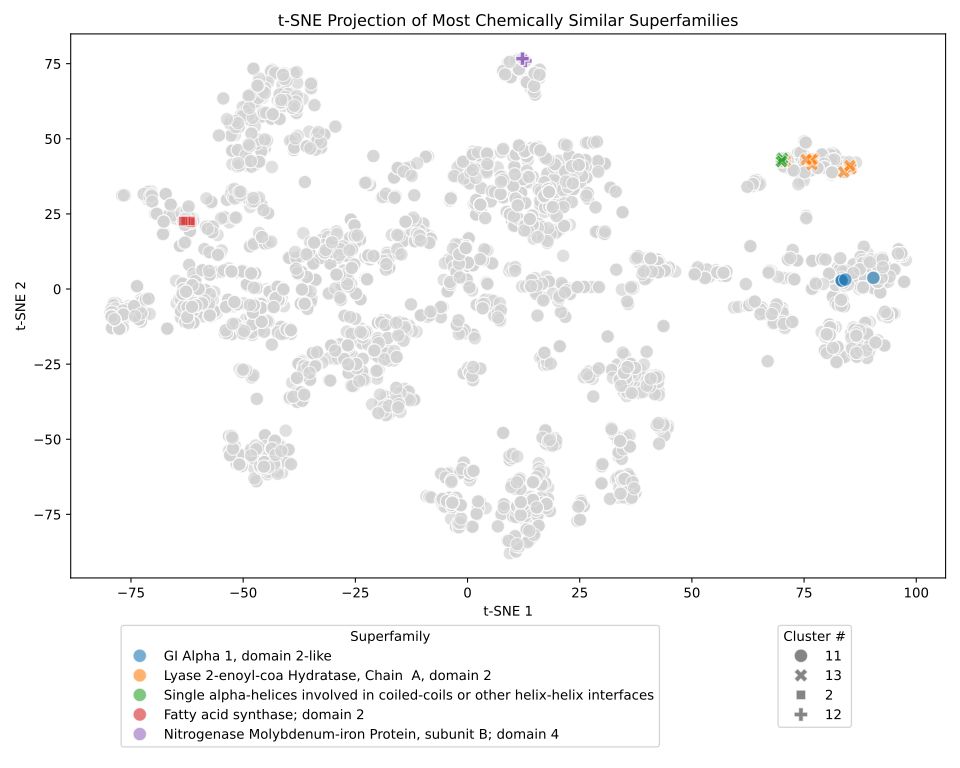
Interestingly if we look at promiscuous superfamilies' (those binding multiple ligands) we find they can fall into "generalised" - binding a wide range of chemically diverse ligands or "specialised" - binding some very chemically similar ligands, categories.
November 14, 2024 at 7:56 PM
Interestingly if we look at promiscuous superfamilies' (those binding multiple ligands) we find they can fall into "generalised" - binding a wide range of chemically diverse ligands or "specialised" - binding some very chemically similar ligands, categories.
Reposted by Rafael Najmanovich
Our latest work has been published in #Bioinformatics Advances ProCogGraph, the spiritual successor of PROCOGNATE. This graph database provides a linkage between proteins domains (SCOP/CATH/Pfam) and cognate ligands for #enzymes in the #PDB. academic.oup.com/bioinformati...
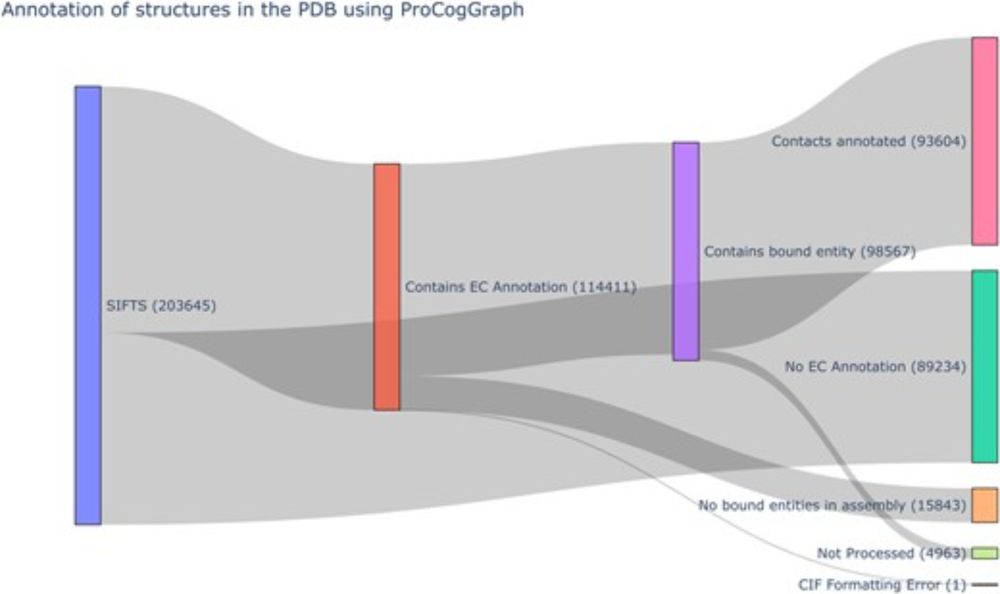
ProCogGraph: a graph-based mapping of cognate ligand domain interactions
AbstractMotivation. Mappings of domain-cognate ligand interactions can enhance our understanding of the core concepts of evolution and be used to aid docki
academic.oup.com
November 14, 2024 at 7:56 PM
Our latest work has been published in #Bioinformatics Advances ProCogGraph, the spiritual successor of PROCOGNATE. This graph database provides a linkage between proteins domains (SCOP/CATH/Pfam) and cognate ligands for #enzymes in the #PDB. academic.oup.com/bioinformati...
Reposted by Rafael Najmanovich

Editorial at The Lancet today on this matter
www.thelancet.com/journals/lan...
www.thelancet.com/journals/lan...

November 14, 2024 at 11:53 PM
Editorial at The Lancet today on this matter
www.thelancet.com/journals/lan...
www.thelancet.com/journals/lan...
Is there a way to search bluesky profile descriptions? That would help to populate my network of who to follow. Alternatively, please suggest who should I follow (including yourself) if you think it fits my research interests (structural/computational biology, design (drugs, proteins), dynamics) etc
November 15, 2024 at 1:55 AM
Is there a way to search bluesky profile descriptions? That would help to populate my network of who to follow. Alternatively, please suggest who should I follow (including yourself) if you think it fits my research interests (structural/computational biology, design (drugs, proteins), dynamics) etc
Happy to share our most recent publication. We use docking and the calculation of NMA-based dynamical signatures to predict the efficiency of mu-Opioid receptor ligands as agonists or antagonists - an example of 3D-QDAR, quantitative dynamics activity relationships. pubs.acs.org/doi/10.1021/...

November 14, 2024 at 1:37 AM
Happy to share our most recent publication. We use docking and the calculation of NMA-based dynamical signatures to predict the efficiency of mu-Opioid receptor ligands as agonists or antagonists - an example of 3D-QDAR, quantitative dynamics activity relationships. pubs.acs.org/doi/10.1021/...