




Noam Teyssier
@noamteyssier.bsky.social
Bioinformatics Scientist at the Arc Institute.
Working at the intersection of functional genomics, systems biology, and machine learning. I also build rusty bioinformatics tools
https://github.com/noamteyssier
Working at the intersection of functional genomics, systems biology, and machine learning. I also build rusty bioinformatics tools
https://github.com/noamteyssier
Reposted by Noam Teyssier

After a long review process, I'm excited that our paper is finally in print: www.cell.com/cell/fulltex...
TL;DR: We use CRISPR screens in iPSC-derived neurons to find a new tau E3 ligase and a relationship between oxidative stress, the proteasome, and tau proteolytic fragments.
More below 👇
TL;DR: We use CRISPR screens in iPSC-derived neurons to find a new tau E3 ligase and a relationship between oxidative stress, the proteasome, and tau proteolytic fragments.
More below 👇

CRISPR screens in iPSC-derived neurons reveal principles of tau proteostasis
CRISPR screens in iPSC-derived neurons reveal that the E3 ubiquitin ligase CRL5SOCS4
ubiquitinates tau, that CUL5 expression is correlated with resilience in human Alzheimer’s
disease, and that electr...
www.cell.com
January 28, 2026 at 5:12 PM
After a long review process, I'm excited that our paper is finally in print: www.cell.com/cell/fulltex...
TL;DR: We use CRISPR screens in iPSC-derived neurons to find a new tau E3 ligase and a relationship between oxidative stress, the proteasome, and tau proteolytic fragments.
More below 👇
TL;DR: We use CRISPR screens in iPSC-derived neurons to find a new tau E3 ligase and a relationship between oxidative stress, the proteasome, and tau proteolytic fragments.
More below 👇
Reposted by Noam Teyssier

Arc bioinformatics scientists @noamteyssier.bsky.social
and Alex Dobin have just released cyto, an ultra-high throughput processor specifically optimized for
@10xgenomics.bsky.social Flex single-cell data.
We are excited to make this resource open source: www.biorxiv.org/content/10.6...
and Alex Dobin have just released cyto, an ultra-high throughput processor specifically optimized for
@10xgenomics.bsky.social Flex single-cell data.
We are excited to make this resource open source: www.biorxiv.org/content/10.6...

January 22, 2026 at 6:13 PM
Arc bioinformatics scientists @noamteyssier.bsky.social
and Alex Dobin have just released cyto, an ultra-high throughput processor specifically optimized for
@10xgenomics.bsky.social Flex single-cell data.
We are excited to make this resource open source: www.biorxiv.org/content/10.6...
and Alex Dobin have just released cyto, an ultra-high throughput processor specifically optimized for
@10xgenomics.bsky.social Flex single-cell data.
We are excited to make this resource open source: www.biorxiv.org/content/10.6...
Today I’m happy to release cyto, a tool I’ve developed at @arcinstitute.org to dramatically increase our computational throughput with 10x-flex single-cell processing by more than 16X!

cyto: ultra high-throughput processing of 10x-flex single cell sequencing https://www.biorxiv.org/content/10.64898/2026.01.21.700936v1
January 22, 2026 at 5:23 PM
Today I’m happy to release cyto, a tool I’ve developed at @arcinstitute.org to dramatically increase our computational throughput with 10x-flex single-cell processing by more than 16X!
It is the year 2026 - bioinformaticians are still trying to figure out the best way to handle fastq
You find yourself in the middle of a fastq file. You see + and @ all over the place. How do you find the start of the current record?
January 9, 2026 at 7:06 PM
It is the year 2026 - bioinformaticians are still trying to figure out the best way to handle fastq
New feature to bqtools v0.4.14 that I'm stoked on!
One of the limiting factors to adopting BINSEQ is that it's new and not widely supported by existing tools.
`bqtools pipe` addresses this by transparently creating FASTX named-pipes which can be processed normally by existing tools.
One of the limiting factors to adopting BINSEQ is that it's new and not widely supported by existing tools.
`bqtools pipe` addresses this by transparently creating FASTX named-pipes which can be processed normally by existing tools.

December 12, 2025 at 7:28 PM
New feature to bqtools v0.4.14 that I'm stoked on!
One of the limiting factors to adopting BINSEQ is that it's new and not widely supported by existing tools.
`bqtools pipe` addresses this by transparently creating FASTX named-pipes which can be processed normally by existing tools.
One of the limiting factors to adopting BINSEQ is that it's new and not widely supported by existing tools.
`bqtools pipe` addresses this by transparently creating FASTX named-pipes which can be processed normally by existing tools.
Reposted by Noam Teyssier

If you ever need to fuzzy search some DNA, sassy is your tool.
Please spread the word; I think many people just outside my own circle could benefit from this :)
cc @rickbitloo.bsky.social
github.com/RagnarGrootK...
Please spread the word; I think many people just outside my own circle could benefit from this :)
cc @rickbitloo.bsky.social
github.com/RagnarGrootK...
December 10, 2025 at 3:50 PM
If you ever need to fuzzy search some DNA, sassy is your tool.
Please spread the word; I think many people just outside my own circle could benefit from this :)
cc @rickbitloo.bsky.social
github.com/RagnarGrootK...
Please spread the word; I think many people just outside my own circle could benefit from this :)
cc @rickbitloo.bsky.social
github.com/RagnarGrootK...
Some optimization on VBQ with the latest binseq update, especially in lossless mode. Some ways to trim the fat:
1. Reuse zstd decoders for each thread. I was creating a decoder for each vbq block which incurred redundant allocations
2. Zero-copy parsing of blocks, referencing similar to paraseq
1. Reuse zstd decoders for each thread. I was creating a decoder for each vbq block which incurred redundant allocations
2. Zero-copy parsing of blocks, referencing similar to paraseq

December 9, 2025 at 10:12 PM
Some optimization on VBQ with the latest binseq update, especially in lossless mode. Some ways to trim the fat:
1. Reuse zstd decoders for each thread. I was creating a decoder for each vbq block which incurred redundant allocations
2. Zero-copy parsing of blocks, referencing similar to paraseq
1. Reuse zstd decoders for each thread. I was creating a decoder for each vbq block which incurred redundant allocations
2. Zero-copy parsing of blocks, referencing similar to paraseq
Reposted by Noam Teyssier

Excited to announce a new bqtools tutorial on sandbox.bio by @noamteyssier.bsky.social! Learn about the BINSEQ file format, and how it can replace FASTQ files for better data compression and faster parallel processing: sandbox.bio/tutorials/bq...

Efficient sequence analysis with bqtools
Interactive bqtools tutorial: learn to analyse sequence data efficiently with BINSEQ files using a command-line interface in your browser.
sandbox.bio
November 18, 2025 at 8:35 PM
Excited to announce a new bqtools tutorial on sandbox.bio by @noamteyssier.bsky.social! Learn about the BINSEQ file format, and how it can replace FASTQ files for better data compression and faster parallel processing: sandbox.bio/tutorials/bq...
I work with large collections of AnnDatas for single-cell work and got tired of opening notebooks for simple operations. Built a CLI tool to handle some common stuff directly from the terminal.
Quick ops: downsample, concat, pseudobulk, QC, metadata export, etc.
github.com/noamteyssier...
Quick ops: downsample, concat, pseudobulk, QC, metadata export, etc.
github.com/noamteyssier...

GitHub - noamteyssier/anntools: a cli-driven anndata toolkit
a cli-driven anndata toolkit. Contribute to noamteyssier/anntools development by creating an account on GitHub.
github.com
November 18, 2025 at 8:14 PM
I work with large collections of AnnDatas for single-cell work and got tired of opening notebooks for simple operations. Built a CLI tool to handle some common stuff directly from the terminal.
Quick ops: downsample, concat, pseudobulk, QC, metadata export, etc.
github.com/noamteyssier...
Quick ops: downsample, concat, pseudobulk, QC, metadata export, etc.
github.com/noamteyssier...
BINSEQ is a high-performance format for sequencing data and bqtools is a CLI tool that lets you create and manipulate these files in the style of samtools.
Excited to release a tutorial with @robert.bio showcasing how to use it to encode, decode, and grep sequences in the browser on sandbox.bio!
Excited to release a tutorial with @robert.bio showcasing how to use it to encode, decode, and grep sequences in the browser on sandbox.bio!

Efficient sequence analysis with bqtools
Interactive bqtools tutorial: learn to analyse sequence data efficiently with BINSEQ files using a command-line interface in your browser.
sandbox.bio
November 14, 2025 at 6:12 PM
BINSEQ is a high-performance format for sequencing data and bqtools is a CLI tool that lets you create and manipulate these files in the style of samtools.
Excited to release a tutorial with @robert.bio showcasing how to use it to encode, decode, and grep sequences in the browser on sandbox.bio!
Excited to release a tutorial with @robert.bio showcasing how to use it to encode, decode, and grep sequences in the browser on sandbox.bio!
New bqtools release with some nice new features!
1. Support for fuzzy matching using sassy
2. Multi-Pattern counting (like `grep -c` but the count is for each individual pattern provided)
3. Pattern files (providing large lists of patterns as either regex or literals)
github.com/ArcInstitute...
1. Support for fuzzy matching using sassy
2. Multi-Pattern counting (like `grep -c` but the count is for each individual pattern provided)
3. Pattern files (providing large lists of patterns as either regex or literals)
github.com/ArcInstitute...

Release bqtools-0.4.8 · ArcInstitute/bqtools
What's Changed
116 support fuzzy grep with sassy by @noamteyssier in #118
119 gate fuzzy matching behind feature flag by @noamteyssier in #120
58 implement a pattern count feature by @noamteyssier...
github.com
November 7, 2025 at 1:12 AM
New bqtools release with some nice new features!
1. Support for fuzzy matching using sassy
2. Multi-Pattern counting (like `grep -c` but the count is for each individual pattern provided)
3. Pattern files (providing large lists of patterns as either regex or literals)
github.com/ArcInstitute...
1. Support for fuzzy matching using sassy
2. Multi-Pattern counting (like `grep -c` but the count is for each individual pattern provided)
3. Pattern files (providing large lists of patterns as either regex or literals)
github.com/ArcInstitute...
I've updated the BINSEQ manuscript to stay up to date with changes since I originally put it out at the beginning of the year
Some notable changes:
1. Support for ambiguous bases with 4bit encoding
2. Support for sequence headers
3. Improved API
www.biorxiv.org/content/10.1...
Some notable changes:
1. Support for ambiguous bases with 4bit encoding
2. Support for sequence headers
3. Improved API
www.biorxiv.org/content/10.1...

BINSEQ: A Family of High-Performance Binary Formats for Nucleotide Sequences
Modern genomics produces billions of sequencing records per run, which are typically stored as gzip-compressed FASTQ files. While this format is widely used, it is not optimal for high-throughput proc...
www.biorxiv.org
October 29, 2025 at 8:41 PM
I've updated the BINSEQ manuscript to stay up to date with changes since I originally put it out at the beginning of the year
Some notable changes:
1. Support for ambiguous bases with 4bit encoding
2. Support for sequence headers
3. Improved API
www.biorxiv.org/content/10.1...
Some notable changes:
1. Support for ambiguous bases with 4bit encoding
2. Support for sequence headers
3. Improved API
www.biorxiv.org/content/10.1...
Was just about to submit a revision for a paper and realized that I wouldn't be able to submit my source for the text because it was written with typst.
Such a bummer - moving this over to tex now but damn what a waste of time!
typst is just so much nicer to work with.
Such a bummer - moving this over to tex now but damn what a waste of time!
typst is just so much nicer to work with.
October 28, 2025 at 11:31 PM
Was just about to submit a revision for a paper and realized that I wouldn't be able to submit my source for the text because it was written with typst.
Such a bummer - moving this over to tex now but damn what a waste of time!
typst is just so much nicer to work with.
Such a bummer - moving this over to tex now but damn what a waste of time!
typst is just so much nicer to work with.
Reposted by Noam Teyssier

Around 10% of your Nanopore reads (SQK-RBK114) are incorrectly trimmed. Here is why, and how our new tool Barbell solves it:
www.biorxiv.org/content/10.1...
Want to get started? github.com/rickbeeloo/b...
www.biorxiv.org/content/10.1...
Want to get started? github.com/rickbeeloo/b...
October 23, 2025 at 8:16 PM
Around 10% of your Nanopore reads (SQK-RBK114) are incorrectly trimmed. Here is why, and how our new tool Barbell solves it:
www.biorxiv.org/content/10.1...
Want to get started? github.com/rickbeeloo/b...
www.biorxiv.org/content/10.1...
Want to get started? github.com/rickbeeloo/b...
Reposted by Noam Teyssier

Thank you Alzforum for featuring our new preprint identifying regulators of disease states of #microglia.
Project led by Amanda McQuade, computation by Reet Mishra, collaboration with the Nunez and De Jager labs.
Alzforum
www.alzforum.org/news/researc...
Preprint
www.biorxiv.org/content/10.1...
Project led by Amanda McQuade, computation by Reet Mishra, collaboration with the Nunez and De Jager labs.
Alzforum
www.alzforum.org/news/researc...
Preprint
www.biorxiv.org/content/10.1...

October 22, 2025 at 6:28 PM
Thank you Alzforum for featuring our new preprint identifying regulators of disease states of #microglia.
Project led by Amanda McQuade, computation by Reet Mishra, collaboration with the Nunez and De Jager labs.
Alzforum
www.alzforum.org/news/researc...
Preprint
www.biorxiv.org/content/10.1...
Project led by Amanda McQuade, computation by Reet Mishra, collaboration with the Nunez and De Jager labs.
Alzforum
www.alzforum.org/news/researc...
Preprint
www.biorxiv.org/content/10.1...
Had an old tool called `hist` to run `sort | uniq -c` years ago but thought up a high-perf impl for it today. Tried it out and found a 25x throughput over the coreutils version.
Big takeaway - arena allocators, hashmaps, and serde work super well together.
github.com/noamteyssier...
Big takeaway - arena allocators, hashmaps, and serde work super well together.
github.com/noamteyssier...

GitHub - noamteyssier/hist-rs: An efficient unique-line counter (25x over `sort | uniq -c`)
An efficient unique-line counter (25x over `sort | uniq -c`) - noamteyssier/hist-rs
github.com
October 22, 2025 at 11:03 PM
Had an old tool called `hist` to run `sort | uniq -c` years ago but thought up a high-perf impl for it today. Tried it out and found a 25x throughput over the coreutils version.
Big takeaway - arena allocators, hashmaps, and serde work super well together.
github.com/noamteyssier...
Big takeaway - arena allocators, hashmaps, and serde work super well together.
github.com/noamteyssier...
Reposted by Noam Teyssier
Paraseq 0.4 is out now! With double the throughput for processing paired-end input :)
github.com/noamteyssier...
github.com/noamteyssier...
September 4, 2025 at 10:41 PM
Paraseq 0.4 is out now! With double the throughput for processing paired-end input :)
github.com/noamteyssier...
github.com/noamteyssier...
Added a feature to bqtools yesterday for colored grep output. Also supports colored FASTX output as well. Already useful this morning as I troubleshoot some sequencing outputs!


September 4, 2025 at 5:56 PM
Added a feature to bqtools yesterday for colored grep output. Also supports colored FASTX output as well. Already useful this morning as I troubleshoot some sequencing outputs!
Reposted by Noam Teyssier
Excited that the paper presenting our mouse brain in vivo CRISPR screening platform is out today in @natneuro.nature.com!
Great team effort, led by Biswa Ramani and @ivlrose.bsky.social in the Kampmann lab.
www.nature.com/articles/s41...
Great team effort, led by Biswa Ramani and @ivlrose.bsky.social in the Kampmann lab.
www.nature.com/articles/s41...

CRISPR screening by AAV episome-sequencing (CrAAVe-seq): a scalable cell-type-specific in vivo platform uncovers neuronal essential genes - Nature Neuroscience
The authors developed an adeno-associated virus-based high-throughput in vivo CRISPR screening platform for endogenous mouse brain cell types. Using this platform, they define genes and pathways essen...
www.nature.com
August 22, 2025 at 10:15 PM
Excited that the paper presenting our mouse brain in vivo CRISPR screening platform is out today in @natneuro.nature.com!
Great team effort, led by Biswa Ramani and @ivlrose.bsky.social in the Kampmann lab.
www.nature.com/articles/s41...
Great team effort, led by Biswa Ramani and @ivlrose.bsky.social in the Kampmann lab.
www.nature.com/articles/s41...
Reposted by Noam Teyssier

Preprint alert!
We present K2Rmini, an ultra-fast, grep-like tool that extracts sequences of interest from FASTA/FASTQ files based on their k-mer content.
www.biorxiv.org/content/10.1...
A thread
We present K2Rmini, an ultra-fast, grep-like tool that extracts sequences of interest from FASTA/FASTQ files based on their k-mer content.
www.biorxiv.org/content/10.1...
A thread

Accelerating k-mer-based sequence filtering
The exponential growth of global sequencing data repositories presents both analytical challenges and opportunities. While k - mer-based indexing has improved scalability over traditional alignment fo...
www.biorxiv.org
July 2, 2025 at 1:00 PM
Preprint alert!
We present K2Rmini, an ultra-fast, grep-like tool that extracts sequences of interest from FASTA/FASTQ files based on their k-mer content.
www.biorxiv.org/content/10.1...
A thread
We present K2Rmini, an ultra-fast, grep-like tool that extracts sequences of interest from FASTA/FASTQ files based on their k-mer content.
www.biorxiv.org/content/10.1...
A thread
Writing in rust again after a long stretch of python is such a breath of fresh air.
June 26, 2025 at 2:47 AM
Writing in rust again after a long stretch of python is such a breath of fresh air.
Reposted by Noam Teyssier
Introducing Arc Institute’s first virtual cell model: STATE

June 23, 2025 at 5:28 PM
Introducing Arc Institute’s first virtual cell model: STATE
Pretty cool little utility and blog post - fun to see the business/pleasure index for rust crates
boydkane.com/projects/cra...
boydkane.com/projects/cra...

Downloaded more for business, or pleasure?
This mini-project was inspired by this tweet: After which I spent about two hours making a small script that grabs data from the rust package repository crates.io, and analyses the ...
boydkane.com
June 18, 2025 at 8:32 PM
Pretty cool little utility and blog post - fun to see the business/pleasure index for rust crates
boydkane.com/projects/cra...
boydkane.com/projects/cra...
Reposted by Noam Teyssier
Preprint on "Improving spliced alignment by modeling splice sites with deep learning". It describes minisplice for modeling splice signals. Minimap2 and miniprot now optionally use the predicted scores to improve spliced alignment.
arxiv.org/abs/2506.12986
arxiv.org/abs/2506.12986
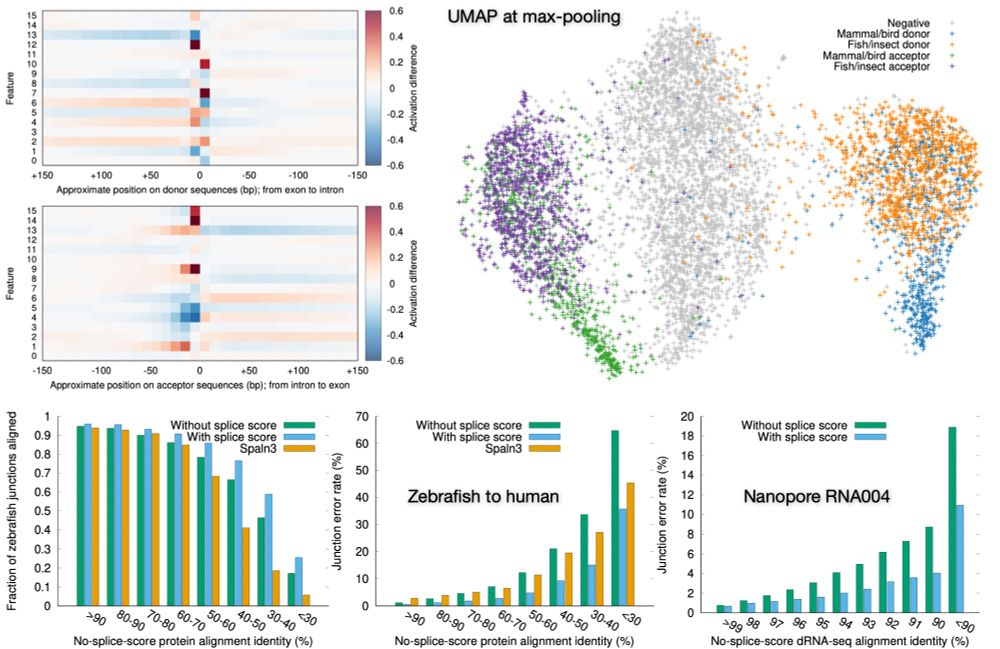
June 17, 2025 at 1:49 AM
Preprint on "Improving spliced alignment by modeling splice sites with deep learning". It describes minisplice for modeling splice signals. Minimap2 and miniprot now optionally use the predicted scores to improve spliced alignment.
arxiv.org/abs/2506.12986
arxiv.org/abs/2506.12986
Reposted by Noam Teyssier

New preprint! Deacon is a versatile tool for filtering FASTA/FASTQ files and streams at hundreds of megabases per second using minimizers, built with rapid metagenomic host depletion in mind, but equally useful for search.
github.com/bede/deacon
github.com/bede/deacon
Deacon: fast sequence filtering and contaminant depletion https://www.biorxiv.org/content/10.1101/2025.06.09.658732v1
June 13, 2025 at 1:25 PM
New preprint! Deacon is a versatile tool for filtering FASTA/FASTQ files and streams at hundreds of megabases per second using minimizers, built with rapid metagenomic host depletion in mind, but equally useful for search.
github.com/bede/deacon
github.com/bede/deacon