



Alexey Nesvizhskii
@nesvilab.bsky.social
Godfrey D. Stobbe Professor of Bioinformatics at U of Michigan. Trained as a theoretical physicist, now focusing on proteomics and proteogenomics. https://fragpipe.nesvilab.org/
Pinned

Exciting news: We have released #FragPipe 23, and it's one of our biggest updates ever. Windows installer, support for TMT on Astral and timsTOF, TMT35, PTM site reports for DIA, improved Astral data handling in #MSFragger, improved diaTracer for diaPASEF data, better Skyline integration, and more!
Reposted by Alexey Nesvizhskii

So glad to see this online today! With FragPipe and the many other Koina APIs, we wanted to democratize deep learning, especially for those without access to expensive GPUs. Major kudos to my co first author Ludwig for setting up the server. I encourage ML developers to put their models on Koina
Exited to share our latest work! Out now in @natcomms.nature.com
Koina aims to transform how #proteomics uses machine learning. You no longer need to be a tech wizard to use ML and now can easily run #ML models. Integrated with FragPipe, Skyline and EncyclopeDIA!
www.nature.com/articles/s41...
Koina aims to transform how #proteomics uses machine learning. You no longer need to be a tech wizard to use ML and now can easily run #ML models. Integrated with FragPipe, Skyline and EncyclopeDIA!
www.nature.com/articles/s41...

Koina: Democratizing machine learning for proteomics research - Nature Communications
Koina is an open-source, online platform that simplifies access to machine learning models in proteomics, enabling easier integration into analysis tools and helping researchers adopt and reuse ML mod...
www.nature.com
November 12, 2025 at 4:37 AM
So glad to see this online today! With FragPipe and the many other Koina APIs, we wanted to democratize deep learning, especially for those without access to expensive GPUs. Major kudos to my co first author Ludwig for setting up the server. I encourage ML developers to put their models on Koina
Reposted by Alexey Nesvizhskii

Exited to share our latest work! Out now in @natcomms.nature.com
Koina aims to transform how #proteomics uses machine learning. You no longer need to be a tech wizard to use ML and now can easily run #ML models. Integrated with FragPipe, Skyline and EncyclopeDIA!
www.nature.com/articles/s41...
Koina aims to transform how #proteomics uses machine learning. You no longer need to be a tech wizard to use ML and now can easily run #ML models. Integrated with FragPipe, Skyline and EncyclopeDIA!
www.nature.com/articles/s41...
Koina: Democratizing machine learning for proteomics research - Nature Communications
Koina is an open-source, online platform that simplifies access to machine learning models in proteomics, enabling easier integration into analysis tools and helping researchers adopt and reuse ML mod...
www.nature.com
November 11, 2025 at 8:06 PM
Exited to share our latest work! Out now in @natcomms.nature.com
Koina aims to transform how #proteomics uses machine learning. You no longer need to be a tech wizard to use ML and now can easily run #ML models. Integrated with FragPipe, Skyline and EncyclopeDIA!
www.nature.com/articles/s41...
Koina aims to transform how #proteomics uses machine learning. You no longer need to be a tech wizard to use ML and now can easily run #ML models. Integrated with FragPipe, Skyline and EncyclopeDIA!
www.nature.com/articles/s41...
Reposted by Alexey Nesvizhskii

This project started 5 years ago. It led us to add isotope-labeling support to #FragPipe/#IonQuant. Since then, the tools have grown so much and are now widely used in #Chemoproteomics.
Huge thanks to everyone, and special thanks to @stephanhacker2.bsky.social and @pzanon.bsky.social
Huge thanks to everyone, and special thanks to @stephanhacker2.bsky.social and @pzanon.bsky.social
How can we study target engagement and selectivity of covalent inhibitors? Which electrophilic probes are best suited to study a certain amino acid?
Our study on "Profiling the proteome-wide selectivity of diverse electrophiles" is published in Nature Chemistry.(1/7)
www.nature.com/articles/s41...
Our study on "Profiling the proteome-wide selectivity of diverse electrophiles" is published in Nature Chemistry.(1/7)
www.nature.com/articles/s41...

Profiling the proteome-wide selectivity of diverse electrophiles - Nature Chemistry
Covalent inhibitors are powerful entities in drug discovery. Now the amino acid selectivity and reactivity of a diverse electrophile library have been assessed proteome-wide using an unbiased workflow...
www.nature.com
October 30, 2025 at 2:15 PM
This project started 5 years ago. It led us to add isotope-labeling support to #FragPipe/#IonQuant. Since then, the tools have grown so much and are now widely used in #Chemoproteomics.
Huge thanks to everyone, and special thanks to @stephanhacker2.bsky.social and @pzanon.bsky.social
Huge thanks to everyone, and special thanks to @stephanhacker2.bsky.social and @pzanon.bsky.social
Reposted by Alexey Nesvizhskii

How can we study target engagement and selectivity of covalent inhibitors? Which electrophilic probes are best suited to study a certain amino acid?
Our study on "Profiling the proteome-wide selectivity of diverse electrophiles" is published in Nature Chemistry.(1/7)
www.nature.com/articles/s41...
Our study on "Profiling the proteome-wide selectivity of diverse electrophiles" is published in Nature Chemistry.(1/7)
www.nature.com/articles/s41...
Profiling the proteome-wide selectivity of diverse electrophiles - Nature Chemistry
Covalent inhibitors are powerful entities in drug discovery. Now the amino acid selectivity and reactivity of a diverse electrophile library have been assessed proteome-wide using an unbiased workflow...
www.nature.com
October 30, 2025 at 10:27 AM
How can we study target engagement and selectivity of covalent inhibitors? Which electrophilic probes are best suited to study a certain amino acid?
Our study on "Profiling the proteome-wide selectivity of diverse electrophiles" is published in Nature Chemistry.(1/7)
www.nature.com/articles/s41...
Our study on "Profiling the proteome-wide selectivity of diverse electrophiles" is published in Nature Chemistry.(1/7)
www.nature.com/articles/s41...
Reposted by Alexey Nesvizhskii

This work led by Elena Levi-D'Ancona, a recent PhD graduate in our lab, was our first cover and only possible due to our amazing team and outstanding collaborators, including @nesvilab.bsky.social and Orian Shirihai, and funding from NIDDK, Breakthrough T1D, and the VA! Thank you! 🤗🙌 2/fin

October 8, 2025 at 8:28 PM
This work led by Elena Levi-D'Ancona, a recent PhD graduate in our lab, was our first cover and only possible due to our amazing team and outstanding collaborators, including @nesvilab.bsky.social and Orian Shirihai, and funding from NIDDK, Breakthrough T1D, and the VA! Thank you! 🤗🙌 2/fin
Reposted by Alexey Nesvizhskii

Proteomics Webinar: DIA with FragPipe, DIA-NN, and Skyline
Presenters: Eduard Sabidó and Brendan MacLean
When: Tuesday, September 16, 8am (Pacific Time)
Register Now ... skyline.ms/project/home...
#massspec #proteomics
Presenters: Eduard Sabidó and Brendan MacLean
When: Tuesday, September 16, 8am (Pacific Time)
Register Now ... skyline.ms/project/home...
#massspec #proteomics
Start Page: /home/software/Skyline/events/2025 Webinars/Webinar 26
skyline.ms
September 15, 2025 at 8:26 AM
Proteomics Webinar: DIA with FragPipe, DIA-NN, and Skyline
Presenters: Eduard Sabidó and Brendan MacLean
When: Tuesday, September 16, 8am (Pacific Time)
Register Now ... skyline.ms/project/home...
#massspec #proteomics
Presenters: Eduard Sabidó and Brendan MacLean
When: Tuesday, September 16, 8am (Pacific Time)
Register Now ... skyline.ms/project/home...
#massspec #proteomics
The University of Michigan now blocks the iProX database, which is a part of the PRIDE consortium of mass spec data repositories. All requests to unblock were denied. Any other US universities in a similar situation? There is a lot of valuable MS proteomics data there no longer accessible to us.
September 10, 2025 at 5:44 PM
The University of Michigan now blocks the iProX database, which is a part of the PRIDE consortium of mass spec data repositories. All requests to unblock were denied. Any other US universities in a similar situation? There is a lot of valuable MS proteomics data there no longer accessible to us.
Reposted by Alexey Nesvizhskii
It was a great pleasure to teach #FragPipe at the Biological Proteomics for Beginners workshop at #UCSD, sponsored by Thermo Fisher Scientific. We had a fantastic group of grad students, postdocs, and professors. Yes, I even got to teach UCSD professors how to analyze bottom-up proteomics data 😁


September 10, 2025 at 2:52 PM
Reposted by Alexey Nesvizhskii

New preprint: We isolate peptide–RNA photo-crosslinks with tunable RNA chains from living cells for mass spec. This maps over 4,700 crosslinking sites across 744 proteins and offers the first glimpse into the RNA sequences in crosslinks by MS. Read here: doi.org/10.1101/2025...

Peptide-RNA photo-crosslinks with tunable RNA chain map protein-RNA interfaces
Photo-crosslinking mass spectrometry enables the identification of protein-RNA interactions in living cells, pinpointing interaction interfaces at single-amino acid resolution. However, current isolat...
doi.org
September 9, 2025 at 11:46 AM
New preprint: We isolate peptide–RNA photo-crosslinks with tunable RNA chains from living cells for mass spec. This maps over 4,700 crosslinking sites across 744 proteins and offers the first glimpse into the RNA sequences in crosslinks by MS. Read here: doi.org/10.1101/2025...
Dan Polasky is indeed a perfect teammate, and not only in our lab but also apparently as a … player in Kubb. I also want to use this opportunity to publicly congratulate Dan for being promoted to Research Assistant Professor starting September 1st!

“Champions” of Kubb at Dagstuhl! Dan Polasky is a great coder AND master gamer - perfect teammate! @nesvilab.bsky.social

August 28, 2025 at 9:11 PM
Dan Polasky is indeed a perfect teammate, and not only in our lab but also apparently as a … player in Kubb. I also want to use this opportunity to publicly congratulate Dan for being promoted to Research Assistant Professor starting September 1st!
#MSFragger Open Search has been around for a while now and used by mass spec folks to screen for chemical artifacts and adducts, e.g. in chemoproteomics data. Happy to see it got 'discovered' by a broader community who are now reporting all sort of cool biological PTMs www.nature.com/articles/s41...

Nucleoside diphosphate kinase A (NME1) catalyses its own oligophosphorylation - Nature Chemistry
Our understanding of how post-translational modification—protein phosphorylation—impacts the complexity of eukaryotic signalling pathways is continuously expanding. Now, protein oligophosphorylation h...
www.nature.com
August 20, 2025 at 3:55 PM
#MSFragger Open Search has been around for a while now and used by mass spec folks to screen for chemical artifacts and adducts, e.g. in chemoproteomics data. Happy to see it got 'discovered' by a broader community who are now reporting all sort of cool biological PTMs www.nature.com/articles/s41...
Reposted by Alexey Nesvizhskii

It's now properly published. If you want to easily check important characteristics of your data before diving into complicated statistics, check out PSManalyst.
PSManalyst: A Dashboard for Visual Quality Control of FragPipe Results | Journal of Proteome Research pubs.acs.org/doi/10.1021/...
PSManalyst: A Dashboard for Visual Quality Control of FragPipe Results | Journal of Proteome Research pubs.acs.org/doi/10.1021/...

PSManalyst: A Dashboard for Visual Quality Control of FragPipe Results
FragPipe is recognized as one of the fastest computational platforms in proteomics, making it a practical solution for the rapid quality control of high-throughput sample analyses. Starting with version 23.0, FragPipe introduced the “Generate Summary Report” feature, offering .pdf reports with essential quality control metrics to address the challenge of intuitively assessing large-scale proteomics data. While traditional spreadsheet formats (e.g., tsv files) are accessible, the complexity of the data often limits user-friendly interpretation. To further enhance accessibility, PSManalyst, a Shiny-based R application, was developed to process FragPipe output files (psm.tsv, protein.tsv, and combined_protein.tsv) and provide interactive, code-free data visualization. Users can filter peptide-spectrum matches (PSMs) by quality scores, visualize protease cleavage fingerprints as heatmaps and SeqLogos, and access a range of quality control metrics and representations such as peptide length distributions, ion densities, mass errors, and wordclouds for overrepresented peptides. The tool facilitates seamless switching between PSM and protein data visualization, offering insights into protein abundance discrepancies, samplewise similarity metrics, protein coverage, and contaminants evaluation. PSManalyst leverages several R libraries (lsa, vegan, ggfortify, ggseqlogo, wordcloud2, tidyverse, ggpointdensity, and plotly) and runs on Windows, MacOS, and Linux, requiring only a local R setup and an IDE. The app is available at (https://github.com/41ison/PSManalyst.
pubs.acs.org
August 15, 2025 at 7:45 PM
It's now properly published. If you want to easily check important characteristics of your data before diving into complicated statistics, check out PSManalyst.
PSManalyst: A Dashboard for Visual Quality Control of FragPipe Results | Journal of Proteome Research pubs.acs.org/doi/10.1021/...
PSManalyst: A Dashboard for Visual Quality Control of FragPipe Results | Journal of Proteome Research pubs.acs.org/doi/10.1021/...
Reposted by Alexey Nesvizhskii
Interested in the proteome-wide selectivity of diverse electrophiles?
The proteomics data for our study on this headed by @pzanon.bsky.social are now public on @pride-ebi.bsky.social: www.ebi.ac.uk/pride/archiv...
Full story: chemrxiv.org/engage/chemr...
#ChemBio #ChemSky #ChemicalProteomics
The proteomics data for our study on this headed by @pzanon.bsky.social are now public on @pride-ebi.bsky.social: www.ebi.ac.uk/pride/archiv...
Full story: chemrxiv.org/engage/chemr...
#ChemBio #ChemSky #ChemicalProteomics

Profiling the proteome-wide selectivity of diverse electrophiles
Targeted covalent inhibitors are powerful entities in drug discovery, but their application has so far mainly been limited to addressing cysteine residues. The development of cysteine-directed covalen...
chemrxiv.org
August 12, 2025 at 9:50 AM
Interested in the proteome-wide selectivity of diverse electrophiles?
The proteomics data for our study on this headed by @pzanon.bsky.social are now public on @pride-ebi.bsky.social: www.ebi.ac.uk/pride/archiv...
Full story: chemrxiv.org/engage/chemr...
#ChemBio #ChemSky #ChemicalProteomics
The proteomics data for our study on this headed by @pzanon.bsky.social are now public on @pride-ebi.bsky.social: www.ebi.ac.uk/pride/archiv...
Full story: chemrxiv.org/engage/chemr...
#ChemBio #ChemSky #ChemicalProteomics
Conventional proteomics searches struggle with many modifications and fully open searches may be difficult to interpret. We introduce a "detailed" mass offset search in #MSFragger boosting interpretability and localization especially in complex cases (e.g. FPOP data): www.biorxiv.org/content/10.1...
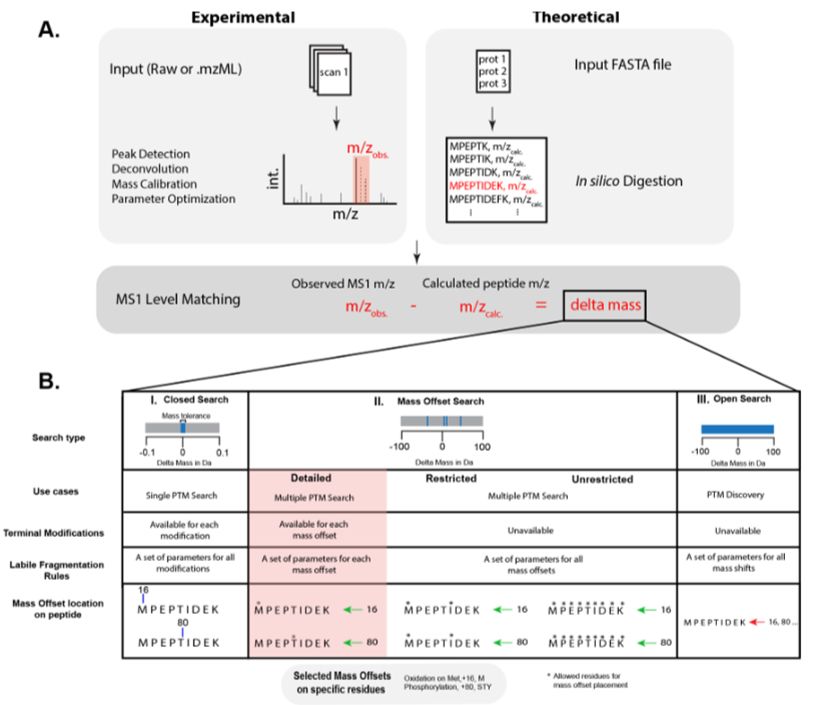
August 1, 2025 at 9:33 PM
Conventional proteomics searches struggle with many modifications and fully open searches may be difficult to interpret. We introduce a "detailed" mass offset search in #MSFragger boosting interpretability and localization especially in complex cases (e.g. FPOP data): www.biorxiv.org/content/10.1...
Reposted by Alexey Nesvizhskii
Type 2 diabetes is often considered a protein misfolding disease. But where are these toxic proteins found? 🤔
🚨In new work out today in @natmetabolism.nature.com, we show that mitochondrial protein misfolding (yes mitos🤯) leads to beta cell damage in T2D. 🚨 nature.com/articles/s42... 1/n
🚨In new work out today in @natmetabolism.nature.com, we show that mitochondrial protein misfolding (yes mitos🤯) leads to beta cell damage in T2D. 🚨 nature.com/articles/s42... 1/n
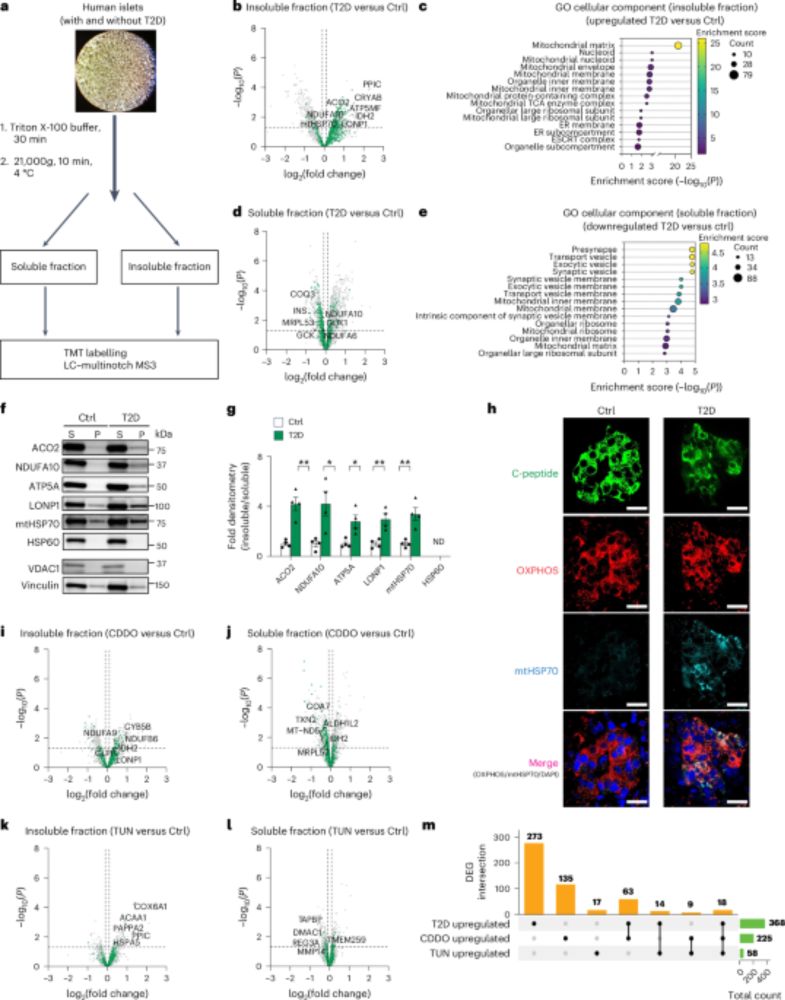
LONP1 regulation of mitochondrial protein folding provides insight into beta cell failure in type 2 diabetes - Nature Metabolism
LONP1, whose expression is downregulated in islets from donors with type 2 diabetes, is vital to mediate efficient mitochondrial protein folding, thus preventing proteotoxicity and promoting islet β c...
nature.com
July 21, 2025 at 10:48 AM
Type 2 diabetes is often considered a protein misfolding disease. But where are these toxic proteins found? 🤔
🚨In new work out today in @natmetabolism.nature.com, we show that mitochondrial protein misfolding (yes mitos🤯) leads to beta cell damage in T2D. 🚨 nature.com/articles/s42... 1/n
🚨In new work out today in @natmetabolism.nature.com, we show that mitochondrial protein misfolding (yes mitos🤯) leads to beta cell damage in T2D. 🚨 nature.com/articles/s42... 1/n
Reposted by Alexey Nesvizhskii

Don't tell PD, but I have quietly switched all of my analyses to Fragpipe. What an extremely powerful software. I'm often amazed at the sheer quantity of information I can get using Fragpipe. Thanks to everyone who recommended it.
Extremely niche troubleshooting request, but is anyone familiar with implementing MSFragger in Proteome Discoverer? A few of us keep seeing these error messages, but none of us are tech savvy enough to decode it. Every time we "solve" it, it happens again.. #proteomics #teammassspec

July 11, 2025 at 8:49 PM
Don't tell PD, but I have quietly switched all of my analyses to Fragpipe. What an extremely powerful software. I'm often amazed at the sheer quantity of information I can get using Fragpipe. Thanks to everyone who recommended it.
Reposted by Alexey Nesvizhskii

DIA, DOA, DUI, DDA, etc. Here is a comparisons of some quantitative proteomics methods from a POV you might not have seen before:
github.com/pwilmart/qua...
github.com/pwilmart/qua...
GitHub - pwilmart/quantitative_proteomics_comparison: Comparison of DIA to spectral counting and TMT quantitative techniques using animal lens studies
Comparison of DIA to spectral counting and TMT quantitative techniques using animal lens studies - pwilmart/quantitative_proteomics_comparison
github.com
July 4, 2025 at 5:53 PM
DIA, DOA, DUI, DDA, etc. Here is a comparisons of some quantitative proteomics methods from a POV you might not have seen before:
github.com/pwilmart/qua...
github.com/pwilmart/qua...
Wow, a record breaking number of the Nesvizhskii lab members attending #ASMS2025! 9 posters, 3 evening workshops, and one Bioinformatics Hub on #FragPipe. Plus multiple collaborative posters with other groups. See you in Baltimore! PS. Below is our recent group photo, including all those attending

May 30, 2025 at 7:45 PM
The power of #MSFragger open search! “we applied the mass-tolerant search engine MSfragger and found that phosphorylation as well as ubiquitination were well preserved after XDNAX. To our great interest, we found an additional modification of 321 Da occurring only in the irradiated SILAC channel”
🚨Our new paper is online🚨
We use zero-distance⚡photo-crosslinking⚡to reveal direct protein-DNA interactions in living cells, enabling quantitative analysis of the DNA-interacting proteome on a timescale of minutes. #DNA #Chromatin #Proteomics
www.cell.com/cell/fulltex...
We use zero-distance⚡photo-crosslinking⚡to reveal direct protein-DNA interactions in living cells, enabling quantitative analysis of the DNA-interacting proteome on a timescale of minutes. #DNA #Chromatin #Proteomics
www.cell.com/cell/fulltex...
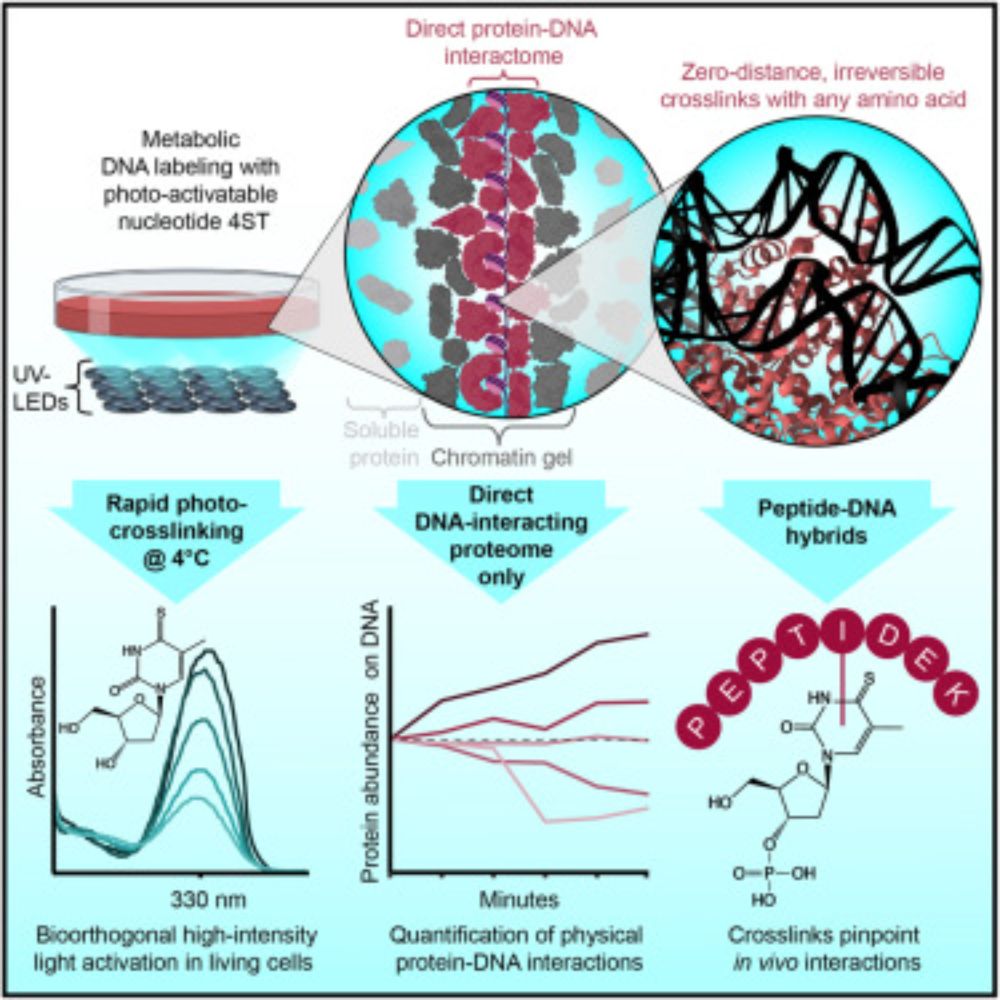
The human proteome with direct physical access to DNA
Zero-distance photo-crosslinking reveals direct protein-DNA interactions in living
cells, enabling quantitative analysis of the DNA-interacting proteome on a timescale
of minutes with single-amino-aci...
www.cell.com
May 23, 2025 at 12:41 PM
The power of #MSFragger open search! “we applied the mass-tolerant search engine MSfragger and found that phosphorylation as well as ubiquitination were well preserved after XDNAX. To our great interest, we found an additional modification of 321 Da occurring only in the irradiated SILAC channel”
Reposted by Alexey Nesvizhskii

Don't miss out! Applications still open for EMBO Practical Course on Targeted proteomics: Advanced tools for biomedical research in Barcelona, Spain, 5 – 10 October 2025
Abstract submission & registration deadline: 15 May
meetings.embo.org/event/25-tar...
#EMBOtargetedProteomics #EMBOevents 🧪
Abstract submission & registration deadline: 15 May
meetings.embo.org/event/25-tar...
#EMBOtargetedProteomics #EMBOevents 🧪

Targeted proteomics: Advanced tools for biomedical research
Targeted proteomics technologies, and specially data-independent acquisition techniques, have revolutionized the landscape of proteomic research in the last decade offering researchers unprecedented …
meetings.embo.org
May 5, 2025 at 9:53 AM
Don't miss out! Applications still open for EMBO Practical Course on Targeted proteomics: Advanced tools for biomedical research in Barcelona, Spain, 5 – 10 October 2025
Abstract submission & registration deadline: 15 May
meetings.embo.org/event/25-tar...
#EMBOtargetedProteomics #EMBOevents 🧪
Abstract submission & registration deadline: 15 May
meetings.embo.org/event/25-tar...
#EMBOtargetedProteomics #EMBOevents 🧪
If there any #Sciex decision makers here on Bluesky - I urge you to reconsider. Skyline/Proteowizard support is not only important for your customers using these tools, but it also benefits other bioinformatics efforts that depend on these tools.

Disappointing to see, across both parties.
Sciex is no longer providing funding for Skyline/Proteowizard as part of the multi-vendor agreement to keep Skyline supported across all major vendors.
I can see the arguments from both sides, just a shame it's resulted in this.
Sciex is no longer providing funding for Skyline/Proteowizard as part of the multi-vendor agreement to keep Skyline supported across all major vendors.
I can see the arguments from both sides, just a shame it's resulted in this.

May 15, 2025 at 1:24 AM
If there any #Sciex decision makers here on Bluesky - I urge you to reconsider. Skyline/Proteowizard support is not only important for your customers using these tools, but it also benefits other bioinformatics efforts that depend on these tools.
Reposted by Alexey Nesvizhskii

Great presentation on #FragPipe and its capabilities. Definitely learned a lot!
Thank you @nesvilab.bsky.social for virtually stopping by to give a talk during our #proteomics users group meeting here @stanford.edu and for answering all our questions 😊
#massspec
#teammassspec
#StanfordProteomics
Thank you @nesvilab.bsky.social for virtually stopping by to give a talk during our #proteomics users group meeting here @stanford.edu and for answering all our questions 😊
#massspec
#teammassspec
#StanfordProteomics

May 8, 2025 at 11:24 PM
Great presentation on #FragPipe and its capabilities. Definitely learned a lot!
Thank you @nesvilab.bsky.social for virtually stopping by to give a talk during our #proteomics users group meeting here @stanford.edu and for answering all our questions 😊
#massspec
#teammassspec
#StanfordProteomics
Thank you @nesvilab.bsky.social for virtually stopping by to give a talk during our #proteomics users group meeting here @stanford.edu and for answering all our questions 😊
#massspec
#teammassspec
#StanfordProteomics
Reposted by Alexey Nesvizhskii
In this release, one of the major improvements is LFQ using IonQuant. Thanks to this excellent preprint (www.biorxiv.org/content/10.1...), we identified and fixed a suboptimal step in the XIC. We're always eager to listen to feedback from the community!

May 7, 2025 at 11:40 PM
In this release, one of the major improvements is LFQ using IonQuant. Thanks to this excellent preprint (www.biorxiv.org/content/10.1...), we identified and fixed a suboptimal step in the XIC. We're always eager to listen to feedback from the community!
Exciting news: We have released #FragPipe 23, and it's one of our biggest updates ever. Windows installer, support for TMT on Astral and timsTOF, TMT35, PTM site reports for DIA, improved Astral data handling in #MSFragger, improved diaTracer for diaPASEF data, better Skyline integration, and more!
May 5, 2025 at 4:06 AM
Exciting news: We have released #FragPipe 23, and it's one of our biggest updates ever. Windows installer, support for TMT on Astral and timsTOF, TMT35, PTM site reports for DIA, improved Astral data handling in #MSFragger, improved diaTracer for diaPASEF data, better Skyline integration, and more!
Are you a fan of FragPipe-Analyst? Or a member of the CPTAC Proteogenomics consortium? Then you for sure know how awesome Leo (Yi Hsiao) is! And today we celebrate him receiving the Rackham Predoctoral Fellowship award from the University of Michigan. Congratulations, Leo!

April 22, 2025 at 8:13 PM
Are you a fan of FragPipe-Analyst? Or a member of the CPTAC Proteogenomics consortium? Then you for sure know how awesome Leo (Yi Hsiao) is! And today we celebrate him receiving the Rackham Predoctoral Fellowship award from the University of Michigan. Congratulations, Leo!