




robbin
@robbinbouwmeester.bsky.social
Postdoc @VIBLifeSciences, @UGent, and @JNJInnovMedEMEA in the @CompOmics group. Interested in Metabolomics, Proteomics, and ML.
Reposted by robbin

Our latest paper introduces new methods for improving peptide retention time predictions in proteomics, incorporating chemical structure information to better handle unseen modifications.
Read all about it in our preprint by👉 doi.org/10.1101/2025...
#Proteomics #AI #MachineLearning
Read all about it in our preprint by👉 doi.org/10.1101/2025...
#Proteomics #AI #MachineLearning

iDeepLC: chemical structure information yields improved retention time prediction of peptides with unseen modifications
Deep learning has notably advanced the field of liquid chromatography–mass spectrometry-based proteomics. Accurate prediction of peptide retention times significantly enhances our ability to match LC-...
doi.org
November 4, 2025 at 8:53 PM
Our latest paper introduces new methods for improving peptide retention time predictions in proteomics, incorporating chemical structure information to better handle unseen modifications.
Read all about it in our preprint by👉 doi.org/10.1101/2025...
#Proteomics #AI #MachineLearning
Read all about it in our preprint by👉 doi.org/10.1101/2025...
#Proteomics #AI #MachineLearning
Reposted by robbin

Exciting news: Preprint on the limitations of current de novo peptide sequencing models on dealing with sequence ambiguity is now out! It focuses on how current models deal with sequence ambiguity, and when and where they go wrong.
Check it out here: www.biorxiv.org/content/10.1...
Check it out here: www.biorxiv.org/content/10.1...

Limitations of de novo sequencing in resolving sequence ambiguity
De novo peptide sequencing enables peptide identification from fragmentation spectra without relying on sequence databases. However, incomplete spectra create ambiguity, making unambiguous identificat...
www.biorxiv.org
August 27, 2025 at 8:15 AM
Exciting news: Preprint on the limitations of current de novo peptide sequencing models on dealing with sequence ambiguity is now out! It focuses on how current models deal with sequence ambiguity, and when and where they go wrong.
Check it out here: www.biorxiv.org/content/10.1...
Check it out here: www.biorxiv.org/content/10.1...
Reposted by robbin

Thanks (especially as I was so vague). It feels like a lot of scripting for sure. After chatting with Magnus, even working through some tutorials like those collabs by @robbinbouwmeester.bsky.social on ProteomicsML wouldn't be a bad idea.
June 25, 2025 at 4:46 PM
Thanks (especially as I was so vague). It feels like a lot of scripting for sure. After chatting with Magnus, even working through some tutorials like those collabs by @robbinbouwmeester.bsky.social on ProteomicsML wouldn't be a bad idea.
Reposted by robbin

And you get the cutest mascotte octopus. His name is Mark! He'll guide you Clippy-wise through your analyses 🙌

June 16, 2025 at 12:32 PM
And you get the cutest mascotte octopus. His name is Mark! He'll guide you Clippy-wise through your analyses 🙌
Reposted by robbin
MLMarker is live! This ML-tool predicts tissue similarity and uncovers biomarkers from your proteomics data. It was trained on public data of healthy human tissues.
Preprint & app: www.biorxiv.org/content/10.1...
Let's chat at #EuPA2025 - Award session (Wednesday) & poster session (Thursday)!
Preprint & app: www.biorxiv.org/content/10.1...
Let's chat at #EuPA2025 - Award session (Wednesday) & poster session (Thursday)!
www.biorxiv.org
June 16, 2025 at 9:31 AM
MLMarker is live! This ML-tool predicts tissue similarity and uncovers biomarkers from your proteomics data. It was trained on public data of healthy human tissues.
Preprint & app: www.biorxiv.org/content/10.1...
Let's chat at #EuPA2025 - Award session (Wednesday) & poster session (Thursday)!
Preprint & app: www.biorxiv.org/content/10.1...
Let's chat at #EuPA2025 - Award session (Wednesday) & poster session (Thursday)!
Reposted by robbin

We recently released a tool to help you with this. 🚀 Say hello to pridepy — your Python for grabbing data from the @pride-ebi.bsky.social!
To search metadata or download files via FTP, Aspera, Globus, or S3, and is perfect for bioinfo workflows.
Check it out 👉 github.com/PRIDE-Archiv...
To search metadata or download files via FTP, Aspera, Globus, or S3, and is perfect for bioinfo workflows.
Check it out 👉 github.com/PRIDE-Archiv...
June 6, 2025 at 8:14 AM
We recently released a tool to help you with this. 🚀 Say hello to pridepy — your Python for grabbing data from the @pride-ebi.bsky.social!
To search metadata or download files via FTP, Aspera, Globus, or S3, and is perfect for bioinfo workflows.
Check it out 👉 github.com/PRIDE-Archiv...
To search metadata or download files via FTP, Aspera, Globus, or S3, and is perfect for bioinfo workflows.
Check it out 👉 github.com/PRIDE-Archiv...
New in DeepLC! Ability to deal with wild, weird, and wobbly LC setups or peptide modifications. This ability is possible with transfer learning; where only a minimal amount of training peptides are needed for accurate retention time predictions.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...

DeepLC introduces transfer learning for accurate LC retention time prediction and adaptation to substantially different modifications and setups
While LC retention time prediction of peptides and their modifications has proven useful, widespread adoption and optimal performance are hindered by variations in experimental parameters. These varia...
www.biorxiv.org
June 4, 2025 at 11:16 AM
New in DeepLC! Ability to deal with wild, weird, and wobbly LC setups or peptide modifications. This ability is possible with transfer learning; where only a minimal amount of training peptides are needed for accurate retention time predictions.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
Reposted by robbin

Fantastic review with an unusual history, growing out of a passionate blog post by @willfondrie.com (willfondrie.com/2024/10/the-...), resulting from a storm (in our teacup) on X during @hupo-org.bsky.social 2024. Great teamwork, authors! pubs.acs.org/doi/10.1021/...
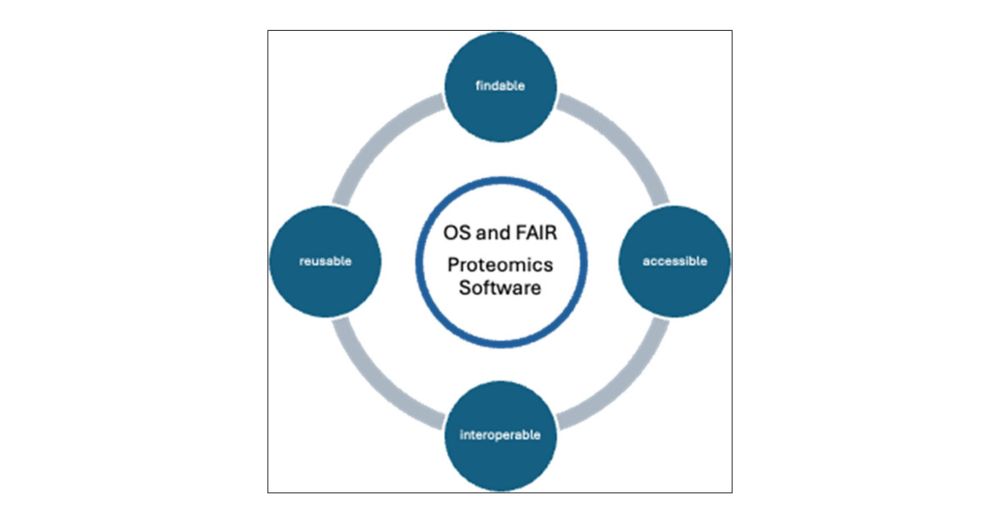
Open-Source and FAIR Research Software for Proteomics
Scientific discovery relies on innovative software as much as experimental methods, especially in proteomics, where computational tools are essential for mass spectrometer setup, data analysis, and interpretation. Since the introduction of SEQUEST, proteomics software has grown into a complex ecosystem of algorithms, predictive models, and workflows, but the field faces challenges, including the increasing complexity of mass spectrometry data, limited reproducibility due to proprietary software, and difficulties integrating with other omics disciplines. Closed-source, platform-specific tools exacerbate these issues by restricting innovation, creating inefficiencies, and imposing hidden costs on the community. Open-source software (OSS), aligned with the FAIR Principles (Findable, Accessible, Interoperable, Reusable), offers a solution by promoting transparency, reproducibility, and community-driven development, which fosters collaboration and continuous improvement. In this manuscript, we explore the role of OSS in computational proteomics, its alignment with FAIR principles, and its potential to address challenges related to licensing, distribution, and standardization. Drawing on lessons from other omics fields, we present a vision for a future where OSS and FAIR principles underpin a transparent, accessible, and innovative proteomics community.
pubs.acs.org
April 24, 2025 at 6:24 PM
Fantastic review with an unusual history, growing out of a passionate blog post by @willfondrie.com (willfondrie.com/2024/10/the-...), resulting from a storm (in our teacup) on X during @hupo-org.bsky.social 2024. Great teamwork, authors! pubs.acs.org/doi/10.1021/...
Reposted by robbin

Classification of Collagens via Peptide Ambiguation, in a Paleoproteomic LC-MS/MS-Based Taxonomic Pipeline #JProteomeRes pubs.acs.org/doi/10.1021/...
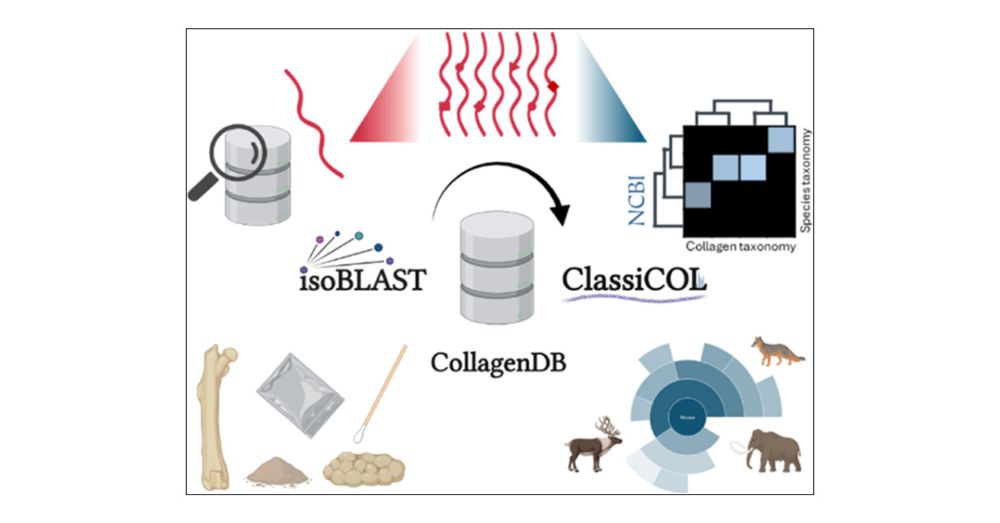
Classification of Collagens via Peptide Ambiguation, in a Paleoproteomic LC-MS/MS-Based Taxonomic Pipeline
Liquid chromatography–mass spectrometry (LC-MS/MS) extends the matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) Zooarcheology by Mass Spectrometry (ZooMS) “mass fingerprinting” approach to species identification by providing fragmentation spectra for each peptide. However, ancient bone samples generate sparse data containing only a few collagen proteins, rendering target–decoy strategies unusable and increasing uncertainty in peptide annotation. To ameliorate this issue, we present a ZooMS/MS data pipeline that builds on a manually curated Collagen database and comprises two novel algorithms: isoBLAST and ClassiCOL. isoBLAST first extends peptide ambiguity by generating all “potential peptide candidates” isobaric to the annotated precursor. The exhaustive set of candidates created is then used to retain or reject different potential paths at each taxonomic branching point from superkingdom to species, until the greatest possible specificity is reached. Uniquely, ClassiCOL allows for the identification of taxonomic mixtures, including contaminated samples, as well as suggesting taxonomies not represented in sequence databases, including extinct taxa. All considered ambiguity is then graphically represented with clear prioritization of the potential taxa in the sample. Using public as well as in-house data acquired on different instruments, we demonstrate the performance of this universal postprocessing and explore the identification of both genetic and sample mixtures. Diet reconstruction from 40,000-year-old cave hyena coprolites illustrates the exciting potential of this approach.
pubs.acs.org
March 14, 2025 at 8:17 AM
Classification of Collagens via Peptide Ambiguation, in a Paleoproteomic LC-MS/MS-Based Taxonomic Pipeline #JProteomeRes pubs.acs.org/doi/10.1021/...
Reposted by robbin

MS2Rescore found its way into #ProteomeDiscoverer 🥳 At least, somewhat, through #MascotServer. Thanks for the implementation and for the nice blog post, @matrixscience.bsky.social!
www.matrixscience.com/blog/using-m...
www.matrixscience.com/blog/using-m...
Using machine learning with Mascot and Proteome Discoverer
www.matrixscience.com
March 7, 2025 at 10:32 AM
MS2Rescore found its way into #ProteomeDiscoverer 🥳 At least, somewhat, through #MascotServer. Thanks for the implementation and for the nice blog post, @matrixscience.bsky.social!
www.matrixscience.com/blog/using-m...
www.matrixscience.com/blog/using-m...
Reposted by robbin

🚀 New preprint alert! We've improved IM2Deep for accurate peptide collisional cross-section (CCS) prediction, even for peptides exhibiting multiple conformations in the gas phase! 🎯
Check it out here:
www.biorxiv.org/content/10.1...
Check it out here:
www.biorxiv.org/content/10.1...

Collisional cross-section prediction for multiconformational peptide ions with IM2Deep
Peptide collisional cross-section (CCS) prediction is complicated by the tendency of peptide ions to exhibit multiple conformations in the gas phase. This adds further complexity to downstream analysi...
www.biorxiv.org
February 23, 2025 at 1:12 PM
🚀 New preprint alert! We've improved IM2Deep for accurate peptide collisional cross-section (CCS) prediction, even for peptides exhibiting multiple conformations in the gas phase! 🎯
Check it out here:
www.biorxiv.org/content/10.1...
Check it out here:
www.biorxiv.org/content/10.1...
Reposted by robbin

DIA-NN 2.0 is released! We consider it the biggest step forward in the history of DIA-NN. On modern LC-MS almost all identifications are now peptidoform-confident, with major improvements e.g. for phospho. Some other cool things too: github.com/vdemichev/Di...

Release DIA-NN 2.0 · vdemichev/DiaNN
We are excited to announce DIA-NN 2.0, the most significant milestone in the history of DIA-NN development.
Key Breakthroughs
Proteoform Confidence mode: DIA-NN 2.0 solves the long-standing chall...
github.com
January 29, 2025 at 9:04 AM
DIA-NN 2.0 is released! We consider it the biggest step forward in the history of DIA-NN. On modern LC-MS almost all identifications are now peptidoform-confident, with major improvements e.g. for phospho. Some other cool things too: github.com/vdemichev/Di...
The end of the year always comes with many 3D print requests. Hope the soon to be PhD will enjoy this ornament :)

December 16, 2024 at 8:53 AM
The end of the year always comes with many 3D print requests. Hope the soon to be PhD will enjoy this ornament :)
Reposted by robbin
Recently, We saw a discussion on the role of open-source in proteomics. Here, experienced developers & researchers maintaining OS tools for years shared this comment to guide newcomers in the field about OS and its role in the field. 💻 #Proteomics #OpenSource chemrxiv.org/engage/chemr...

Open-source and FAIR Research Software for Proteomics
Scientific discovery relies on innovative software as much as experimental methods, especially in proteomics, where computational tools are essential for mass spectrometer setup, data analysis, and in...
chemrxiv.org
December 9, 2024 at 1:03 PM
Recently, We saw a discussion on the role of open-source in proteomics. Here, experienced developers & researchers maintaining OS tools for years shared this comment to guide newcomers in the field about OS and its role in the field. 💻 #Proteomics #OpenSource chemrxiv.org/engage/chemr...
Reposted by robbin

This was a ton of fun to write with @ypriverol.bsky.social and all of the other authors 👏
Our goal was to share a vision of #OSS #proteomics for us to build toward, and propose some ways to get there 🚀
I’m blown away by how many folks contributed and how much it evolved beyond just my voice 🙌
Our goal was to share a vision of #OSS #proteomics for us to build toward, and propose some ways to get there 🚀
I’m blown away by how many folks contributed and how much it evolved beyond just my voice 🙌
Recently, We saw a discussion on the role of open-source in proteomics. Here, experienced developers & researchers maintaining OS tools for years shared this comment to guide newcomers in the field about OS and its role in the field. 💻 #Proteomics #OpenSource chemrxiv.org/engage/chemr...
Open-source and FAIR Research Software for Proteomics
Scientific discovery relies on innovative software as much as experimental methods, especially in proteomics, where computational tools are essential for mass spectrometer setup, data analysis, and in...
chemrxiv.org
December 9, 2024 at 3:59 PM
This was a ton of fun to write with @ypriverol.bsky.social and all of the other authors 👏
Our goal was to share a vision of #OSS #proteomics for us to build toward, and propose some ways to get there 🚀
I’m blown away by how many folks contributed and how much it evolved beyond just my voice 🙌
Our goal was to share a vision of #OSS #proteomics for us to build toward, and propose some ways to get there 🚀
I’m blown away by how many folks contributed and how much it evolved beyond just my voice 🙌
Reposted by robbin
Today, @robbedevr.bsky.social and @carojachmann.bsky.social presented their work on #IM2Deep and #ProteoBench at #BePAc2024.
Learn more at doi.org/10.1101/2024... and proteobench.readthedocs.io.
Learn more at doi.org/10.1101/2024... and proteobench.readthedocs.io.


December 6, 2024 at 2:52 PM
Today, @robbedevr.bsky.social and @carojachmann.bsky.social presented their work on #IM2Deep and #ProteoBench at #BePAc2024.
Learn more at doi.org/10.1101/2024... and proteobench.readthedocs.io.
Learn more at doi.org/10.1101/2024... and proteobench.readthedocs.io.
Reposted by robbin
I think it was Robbin Bouwmeester’s talk at EuBIC where it was SAGE to MS2Rescore and it seemed quietly bad ass. This update is making it look even more attractive as my new fav proteomics pipeline.

MS²Rescore 3.0 is a modular, flexible, and
user-friendly platform to boost peptide
identifications, as showcased with MS Amanda 3.0 chemrxiv.org/engage/...
---
#proteomics #prot-preprint
user-friendly platform to boost peptide
identifications, as showcased with MS Amanda 3.0 chemrxiv.org/engage/...
---
#proteomics #prot-preprint

January 29, 2024 at 12:52 PM
I think it was Robbin Bouwmeester’s talk at EuBIC where it was SAGE to MS2Rescore and it seemed quietly bad ass. This update is making it look even more attractive as my new fav proteomics pipeline.