



Robbe Devreese
@robbedevr.bsky.social
PhD student in computational proteomics
Reposted by Robbe Devreese

We are happy to share that the preprint for Proteobench is available now on biorxiv!
Check it out here: www.biorxiv.org/content/10.6...
Congratulations and thanks to everyone involved for the great effort!
Check it out here: www.biorxiv.org/content/10.6...
Congratulations and thanks to everyone involved for the great effort!

ProteoBench: the community-curated platform for comparing proteomics data analysis workflows
Mass spectrometry (MS)-based proteomics is a well-established strategy for analyzing complex biological mixtures. Many MS instruments and data acquisition strategies are available, and the data they a...
www.biorxiv.org
December 15, 2025 at 8:55 AM
We are happy to share that the preprint for Proteobench is available now on biorxiv!
Check it out here: www.biorxiv.org/content/10.6...
Congratulations and thanks to everyone involved for the great effort!
Check it out here: www.biorxiv.org/content/10.6...
Congratulations and thanks to everyone involved for the great effort!
Reposted by Robbe Devreese

Exited to share our latest work! Out now in @natcomms.nature.com
Koina aims to transform how #proteomics uses machine learning. You no longer need to be a tech wizard to use ML and now can easily run #ML models. Integrated with FragPipe, Skyline and EncyclopeDIA!
www.nature.com/articles/s41...
Koina aims to transform how #proteomics uses machine learning. You no longer need to be a tech wizard to use ML and now can easily run #ML models. Integrated with FragPipe, Skyline and EncyclopeDIA!
www.nature.com/articles/s41...

Koina: Democratizing machine learning for proteomics research - Nature Communications
Koina is an open-source, online platform that simplifies access to machine learning models in proteomics, enabling easier integration into analysis tools and helping researchers adopt and reuse ML mod...
www.nature.com
November 11, 2025 at 8:06 PM
Exited to share our latest work! Out now in @natcomms.nature.com
Koina aims to transform how #proteomics uses machine learning. You no longer need to be a tech wizard to use ML and now can easily run #ML models. Integrated with FragPipe, Skyline and EncyclopeDIA!
www.nature.com/articles/s41...
Koina aims to transform how #proteomics uses machine learning. You no longer need to be a tech wizard to use ML and now can easily run #ML models. Integrated with FragPipe, Skyline and EncyclopeDIA!
www.nature.com/articles/s41...
Reposted by Robbe Devreese

pubs.acs.org/doi/10.1021/... IM2Deep might win best algorithm name of 2025
pubs.acs.org
July 15, 2025 at 7:59 PM
pubs.acs.org/doi/10.1021/... IM2Deep might win best algorithm name of 2025
Reposted by Robbe Devreese
Marie Locard-Paulet presenting the EuBIC-MS #ProteoBench project at the #EuPA2025 conference.
👉 Learn more about our open proteomics software benchmarking platform at proteobench.readthedocs.io.
#Proteomics #Bioinformatics #Benchmarking
👉 Learn more about our open proteomics software benchmarking platform at proteobench.readthedocs.io.
#Proteomics #Bioinformatics #Benchmarking

June 19, 2025 at 12:13 PM
Marie Locard-Paulet presenting the EuBIC-MS #ProteoBench project at the #EuPA2025 conference.
👉 Learn more about our open proteomics software benchmarking platform at proteobench.readthedocs.io.
#Proteomics #Bioinformatics #Benchmarking
👉 Learn more about our open proteomics software benchmarking platform at proteobench.readthedocs.io.
#Proteomics #Bioinformatics #Benchmarking
Reposted by Robbe Devreese

MLMarker is live! This ML-tool predicts tissue similarity and uncovers biomarkers from your proteomics data. It was trained on public data of healthy human tissues.
Preprint & app: www.biorxiv.org/content/10.1...
Let's chat at #EuPA2025 - Award session (Wednesday) & poster session (Thursday)!
Preprint & app: www.biorxiv.org/content/10.1...
Let's chat at #EuPA2025 - Award session (Wednesday) & poster session (Thursday)!
www.biorxiv.org
June 16, 2025 at 9:31 AM
MLMarker is live! This ML-tool predicts tissue similarity and uncovers biomarkers from your proteomics data. It was trained on public data of healthy human tissues.
Preprint & app: www.biorxiv.org/content/10.1...
Let's chat at #EuPA2025 - Award session (Wednesday) & poster session (Thursday)!
Preprint & app: www.biorxiv.org/content/10.1...
Let's chat at #EuPA2025 - Award session (Wednesday) & poster session (Thursday)!
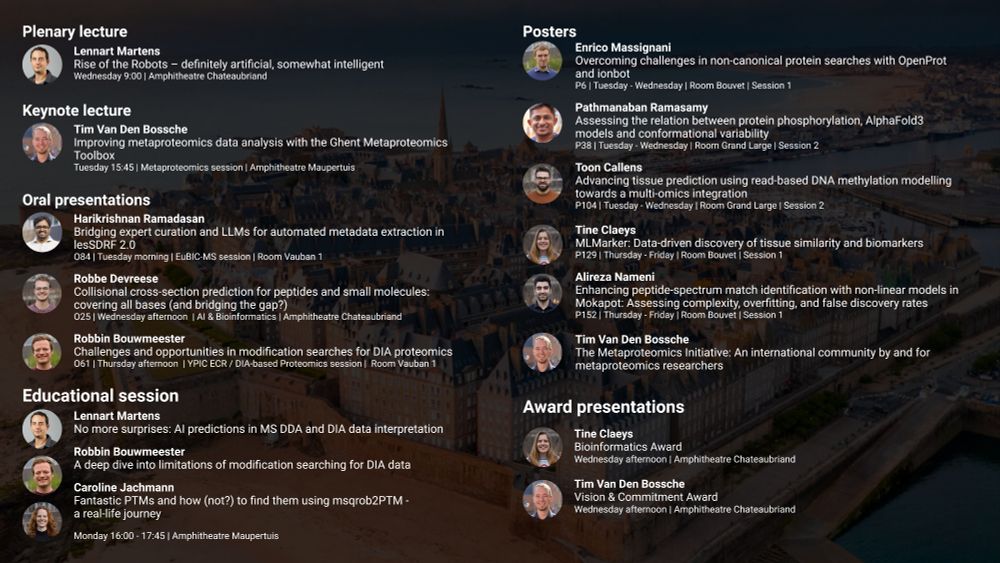
Reposted by Robbe Devreese
Join us in Saint-Malo for the #EuBIC-MS session at #EuPA2025!
After a short introduction, we have three exciting talks lined up, as well as an interactive discussion on open issues in computational proteomics.
@eupaproteomics.bsky.social @uszkoreitju.bsky.social @harirmds.bsky.social
After a short introduction, we have three exciting talks lined up, as well as an interactive discussion on open issues in computational proteomics.
@eupaproteomics.bsky.social @uszkoreitju.bsky.social @harirmds.bsky.social

June 13, 2025 at 1:56 PM
Join us in Saint-Malo for the #EuBIC-MS session at #EuPA2025!
After a short introduction, we have three exciting talks lined up, as well as an interactive discussion on open issues in computational proteomics.
@eupaproteomics.bsky.social @uszkoreitju.bsky.social @harirmds.bsky.social
After a short introduction, we have three exciting talks lined up, as well as an interactive discussion on open issues in computational proteomics.
@eupaproteomics.bsky.social @uszkoreitju.bsky.social @harirmds.bsky.social
Reposted by Robbe Devreese

New in DeepLC! Ability to deal with wild, weird, and wobbly LC setups or peptide modifications. This ability is possible with transfer learning; where only a minimal amount of training peptides are needed for accurate retention time predictions.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...

DeepLC introduces transfer learning for accurate LC retention time prediction and adaptation to substantially different modifications and setups
While LC retention time prediction of peptides and their modifications has proven useful, widespread adoption and optimal performance are hindered by variations in experimental parameters. These varia...
www.biorxiv.org
June 4, 2025 at 11:16 AM
New in DeepLC! Ability to deal with wild, weird, and wobbly LC setups or peptide modifications. This ability is possible with transfer learning; where only a minimal amount of training peptides are needed for accurate retention time predictions.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
Reposted by Robbe Devreese


May 27, 2025 at 5:54 AM
Reposted by Robbe Devreese

What an amazing team, this #proteobench people. It was very inspiring to join, support and push this Proteobench project forward representing Core4life during the Proteobench hackaton this week. Thank you to Institut Pasteur, for hosting the joint adventure!




May 23, 2025 at 7:39 PM
What an amazing team, this #proteobench people. It was very inspiring to join, support and push this Proteobench project forward representing Core4life during the Proteobench hackaton this week. Thank you to Institut Pasteur, for hosting the joint adventure!
Reposted by Robbe Devreese
🚀 New preprint alert! We've improved IM2Deep for accurate peptide collisional cross-section (CCS) prediction, even for peptides exhibiting multiple conformations in the gas phase! 🎯
Check it out here:
www.biorxiv.org/content/10.1...
Check it out here:
www.biorxiv.org/content/10.1...

Collisional cross-section prediction for multiconformational peptide ions with IM2Deep
Peptide collisional cross-section (CCS) prediction is complicated by the tendency of peptide ions to exhibit multiple conformations in the gas phase. This adds further complexity to downstream analysi...
www.biorxiv.org
February 23, 2025 at 1:12 PM
🚀 New preprint alert! We've improved IM2Deep for accurate peptide collisional cross-section (CCS) prediction, even for peptides exhibiting multiple conformations in the gas phase! 🎯
Check it out here:
www.biorxiv.org/content/10.1...
Check it out here:
www.biorxiv.org/content/10.1...
Reposted by Robbe Devreese

Collisional cross-section prediction for multiconformational peptide ions with IM2Deep www.biorxiv.org/cont...
---
#proteomics #prot-preprint
---
#proteomics #prot-preprint

February 23, 2025 at 2:00 PM
Collisional cross-section prediction for multiconformational peptide ions with IM2Deep www.biorxiv.org/cont...
---
#proteomics #prot-preprint
---
#proteomics #prot-preprint
Reposted by Robbe Devreese
What excites me most is that it introduces the first ML-based solution for peptide multiconformers. But that’s not all! We also demonstrate a substantial performance boost for uniconforming peptides.
Our findings are clear: multiconformer peptides cannot be overlooked when predicting CCS!
Our findings are clear: multiconformer peptides cannot be overlooked when predicting CCS!
February 23, 2025 at 1:35 PM
What excites me most is that it introduces the first ML-based solution for peptide multiconformers. But that’s not all! We also demonstrate a substantial performance boost for uniconforming peptides.
Our findings are clear: multiconformer peptides cannot be overlooked when predicting CCS!
Our findings are clear: multiconformer peptides cannot be overlooked when predicting CCS!
🚀 New preprint alert! We've improved IM2Deep for accurate peptide collisional cross-section (CCS) prediction, even for peptides exhibiting multiple conformations in the gas phase! 🎯
Check it out here:
www.biorxiv.org/content/10.1...
Check it out here:
www.biorxiv.org/content/10.1...
Collisional cross-section prediction for multiconformational peptide ions with IM2Deep
Peptide collisional cross-section (CCS) prediction is complicated by the tendency of peptide ions to exhibit multiple conformations in the gas phase. This adds further complexity to downstream analysi...
www.biorxiv.org
February 23, 2025 at 1:12 PM
🚀 New preprint alert! We've improved IM2Deep for accurate peptide collisional cross-section (CCS) prediction, even for peptides exhibiting multiple conformations in the gas phase! 🎯
Check it out here:
www.biorxiv.org/content/10.1...
Check it out here:
www.biorxiv.org/content/10.1...
Seems reasonable to dedicate my first Bluesky post to the following:
Our latest research, TIMS²Rescore, is now published in Journal of Proteome Research! 🎉
Read it here: pubs.acs.org/doi/full/10....
A huge thanks to all our collaborators for making this happen!
Our latest research, TIMS²Rescore, is now published in Journal of Proteome Research! 🎉
Read it here: pubs.acs.org/doi/full/10....
A huge thanks to all our collaborators for making this happen!

TIMS2Rescore: A Data Dependent Acquisition-Parallel Accumulation and Serial Fragmentation-Optimized Data-Driven Rescoring Pipeline Based on MS2Rescore
The high throughput analysis of proteins with mass spectrometry (MS) is highly valuable for understanding human biology, discovering disease biomarkers, identifying therapeutic targets, and exploring pathogen interactions. To achieve these goals, specialized proteomics subfields, including plasma proteomics, immunopeptidomics, and metaproteomics, must tackle specific analytical challenges, such as an increased identification ambiguity compared to routine proteomics experiments. Technical advancements in MS instrumentation can mitigate these issues by acquiring more discerning information at higher sensitivity levels. This is exemplified by the incorporation of ion mobility and parallel accumulation and serial fragmentation (PASEF) technologies in timsTOF instruments. In addition, AI-based bioinformatics solutions can help overcome ambiguity issues by integrating more data into the identification workflow. Here, we introduce TIMS2Rescore, a data-driven rescoring workflow optimized for DDA-PASEF data from timsTOF instruments. This platform includes new timsTOF MS2PIP spectrum prediction models and IM2Deep, a new deep learning-based peptide ion mobility predictor. Furthermore, to fully streamline data throughput, TIMS2Rescore directly accepts Bruker raw mass spectrometry data and search results from ProteoScape and many other search engines, including Sage and PEAKS. We showcase TIMS2Rescore performance on plasma proteomics, immunopeptidomics (HLA class I and II), and metaproteomics data sets. TIMS2Rescore is open-source and freely available at https://github.com/compomics/tims2rescore.
pubs.acs.org
February 7, 2025 at 2:13 PM
Seems reasonable to dedicate my first Bluesky post to the following:
Our latest research, TIMS²Rescore, is now published in Journal of Proteome Research! 🎉
Read it here: pubs.acs.org/doi/full/10....
A huge thanks to all our collaborators for making this happen!
Our latest research, TIMS²Rescore, is now published in Journal of Proteome Research! 🎉
Read it here: pubs.acs.org/doi/full/10....
A huge thanks to all our collaborators for making this happen!
Reposted by Robbe Devreese

Today, @robbedevr.bsky.social and @carojachmann.bsky.social presented their work on #IM2Deep and #ProteoBench at #BePAc2024.
Learn more at doi.org/10.1101/2024... and proteobench.readthedocs.io.
Learn more at doi.org/10.1101/2024... and proteobench.readthedocs.io.


December 6, 2024 at 2:52 PM
Today, @robbedevr.bsky.social and @carojachmann.bsky.social presented their work on #IM2Deep and #ProteoBench at #BePAc2024.
Learn more at doi.org/10.1101/2024... and proteobench.readthedocs.io.
Learn more at doi.org/10.1101/2024... and proteobench.readthedocs.io.