



Tijs van Lieshout
@magiduck.bsky.social
PhD student within the Functional Genomics group (Franke lab), University Medical Centre Groningen. Interested in gene networks, sequence-based models and non-coding somatic mutations
Pinned

We are excited to share our manuscript “Identification of (ultra-)rare functional promoter mutations in cancer using sequence-based deep learning models” (www.medrxiv.org/content/10.1...). (1/16) 🧵

Reposted by Tijs van Lieshout

We are excited to share GPN-Star, a cost-effective, biologically grounded genomic language modeling framework that achieves state-of-the-art performance across a wide range of variant effect prediction tasks relevant to human genetics.
www.biorxiv.org/content/10.1...
(1/n)
www.biorxiv.org/content/10.1...
(1/n)

September 22, 2025 at 5:29 AM
We are excited to share GPN-Star, a cost-effective, biologically grounded genomic language modeling framework that achieves state-of-the-art performance across a wide range of variant effect prediction tasks relevant to human genetics.
www.biorxiv.org/content/10.1...
(1/n)
www.biorxiv.org/content/10.1...
(1/n)
Reposted by Tijs van Lieshout

Excited for a major milestone in our efforts to map enhancers and interpret variants in the human genome:
The E2G Portal! e2g.stanford.edu
This collates our predictions of enhancer-gene regulatory interactions across >1,600 cell types and tissues.
Uses cases 👇
1/
The E2G Portal! e2g.stanford.edu
This collates our predictions of enhancer-gene regulatory interactions across >1,600 cell types and tissues.
Uses cases 👇
1/
September 18, 2025 at 4:14 PM
Excited for a major milestone in our efforts to map enhancers and interpret variants in the human genome:
The E2G Portal! e2g.stanford.edu
This collates our predictions of enhancer-gene regulatory interactions across >1,600 cell types and tissues.
Uses cases 👇
1/
The E2G Portal! e2g.stanford.edu
This collates our predictions of enhancer-gene regulatory interactions across >1,600 cell types and tissues.
Uses cases 👇
1/
Reposted by Tijs van Lieshout

An assessment of DNA language models concludes:
◼️ They do not offer compelling gains over baseline models
Their performance is inconsistent and requires much more compute.
arxiv.org/abs/2412.05430
◼️ They do not offer compelling gains over baseline models
Their performance is inconsistent and requires much more compute.
arxiv.org/abs/2412.05430

June 23, 2025 at 8:21 PM
An assessment of DNA language models concludes:
◼️ They do not offer compelling gains over baseline models
Their performance is inconsistent and requires much more compute.
arxiv.org/abs/2412.05430
◼️ They do not offer compelling gains over baseline models
Their performance is inconsistent and requires much more compute.
arxiv.org/abs/2412.05430
Reposted by Tijs van Lieshout

I'm only partially kidding cuz he definitely is forcing me to modify some of my slides 😂. But I did want to express some thoughts on the lightweight specialized single task (ST) models vs kitchensink multi task (MT) model paradigms a bit. 2/
June 26, 2025 at 6:29 AM
I'm only partially kidding cuz he definitely is forcing me to modify some of my slides 😂. But I did want to express some thoughts on the lightweight specialized single task (ST) models vs kitchensink multi task (MT) model paradigms a bit. 2/
Reposted by Tijs van Lieshout
This a really exciting leap forward for genomic sequence to activity gene regulation models. It is a genuine improvement over pretty much all SOTA models spanning a wide range of regulatory, transcriptional and post-transcriptional processes. 1/

Excited to launch our AlphaGenome API goo.gle/3ZPUeFX along with the preprint goo.gle/45AkUyc describing and evaluating our latest DNA sequence model powering the API. Looking forward to seeing how scientists use it! @googledeepmind
June 25, 2025 at 4:18 PM
This a really exciting leap forward for genomic sequence to activity gene regulation models. It is a genuine improvement over pretty much all SOTA models spanning a wide range of regulatory, transcriptional and post-transcriptional processes. 1/
Reposted by Tijs van Lieshout
One thing that really bothers me with the new "virtual cell" terminology is that it is currently largely focused on a very narrow definition of models that can predict effects of trans perturbations (gene dosage, drugs etc) on gene expression. 1/
June 28, 2025 at 10:38 AM
One thing that really bothers me with the new "virtual cell" terminology is that it is currently largely focused on a very narrow definition of models that can predict effects of trans perturbations (gene dosage, drugs etc) on gene expression. 1/
Reposted by Tijs van Lieshout

1/ DNA sequence models like Borzoi predict gene expression and variant effects across tissues — but how can someone adapt the model to a custom experiment? @drkbio.bsky.social, Johannes Linder and I propose a solution via parameter-efficient fine-tuning (PEFT).
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...

Parameter-Efficient Fine-Tuning of a Supervised Regulatory Sequence Model
DNA sequence deep learning models accurately predict epigenetic and transcriptional profiles, enabling analysis of gene regulation and genetic variant effects. While large-scale training models like E...
www.biorxiv.org
June 2, 2025 at 8:09 PM
1/ DNA sequence models like Borzoi predict gene expression and variant effects across tissues — but how can someone adapt the model to a custom experiment? @drkbio.bsky.social, Johannes Linder and I propose a solution via parameter-efficient fine-tuning (PEFT).
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
Reposted by Tijs van Lieshout

Many of you enjoy our sequence-based model of single-cell RNA and ATAC data scooby... Don't miss Laura Marten's talk at the upcoming Kipoi seminar about it this Wed!
@lauradmartens.bsky.social @johahi.bsky.social @kipoizoo.bsky.social
Last preprint version:
www.biorxiv.org/content/10.1...
@lauradmartens.bsky.social @johahi.bsky.social @kipoizoo.bsky.social
Last preprint version:
www.biorxiv.org/content/10.1...
May 5, 2025 at 4:26 PM
Many of you enjoy our sequence-based model of single-cell RNA and ATAC data scooby... Don't miss Laura Marten's talk at the upcoming Kipoi seminar about it this Wed!
@lauradmartens.bsky.social @johahi.bsky.social @kipoizoo.bsky.social
Last preprint version:
www.biorxiv.org/content/10.1...
@lauradmartens.bsky.social @johahi.bsky.social @kipoizoo.bsky.social
Last preprint version:
www.biorxiv.org/content/10.1...
Reposted by Tijs van Lieshout

We studied non-coding somatic mutations in WGS data of 24,000 cancer patients. We employed sequence-based models to identify promotors, enriched for somatic variants that are predicted to affect expression. A great collaboration with the teams of Emile Voest, Bas van Steensel and Jeroen de Ridder!
We are excited to share our manuscript “Identification of (ultra-)rare functional promoter mutations in cancer using sequence-based deep learning models” (www.medrxiv.org/content/10.1...). (1/16) 🧵
May 7, 2025 at 8:03 AM
We studied non-coding somatic mutations in WGS data of 24,000 cancer patients. We employed sequence-based models to identify promotors, enriched for somatic variants that are predicted to affect expression. A great collaboration with the teams of Emile Voest, Bas van Steensel and Jeroen de Ridder!
Reposted by Tijs van Lieshout

Identification of (ultra-)rare functional promoter mutations in cancer using sequence-based deep learning models https://www.medrxiv.org/content/10.1101/2025.05.06.25327057v1
May 7, 2025 at 3:40 AM
Identification of (ultra-)rare functional promoter mutations in cancer using sequence-based deep learning models https://www.medrxiv.org/content/10.1101/2025.05.06.25327057v1
We are excited to share our manuscript “Identification of (ultra-)rare functional promoter mutations in cancer using sequence-based deep learning models” (www.medrxiv.org/content/10.1...). (1/16) 🧵
May 7, 2025 at 7:04 AM
We are excited to share our manuscript “Identification of (ultra-)rare functional promoter mutations in cancer using sequence-based deep learning models” (www.medrxiv.org/content/10.1...). (1/16) 🧵
Reposted by Tijs van Lieshout

As someone who has reported on AI for 7 years and covered China tech as well, I think the biggest lesson to be drawn from DeepSeek is the huge cracks it illustrates with the current dominant paradigm of AI development. A long thread. 1/
January 27, 2025 at 2:12 PM
As someone who has reported on AI for 7 years and covered China tech as well, I think the biggest lesson to be drawn from DeepSeek is the huge cracks it illustrates with the current dominant paradigm of AI development. A long thread. 1/
Reposted by Tijs van Lieshout

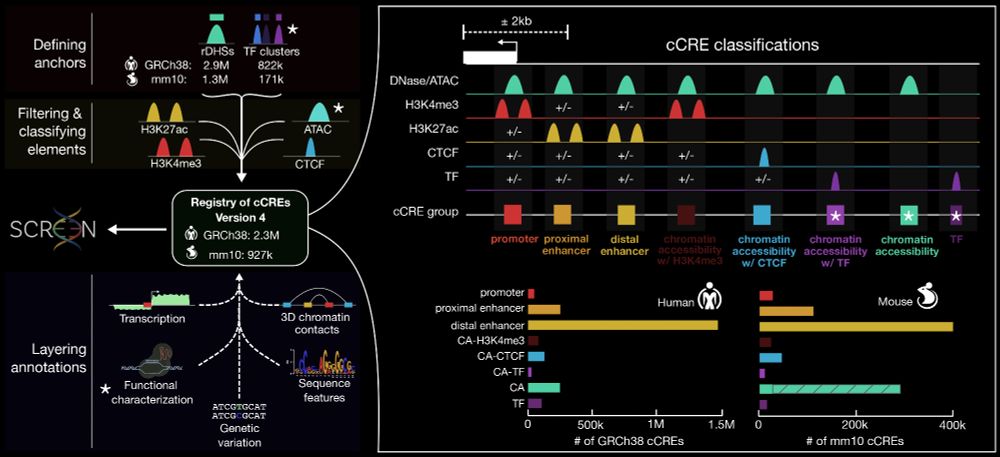
Now available as preprint:
The ENCODE 4 expanded registry of regulatory elements
- 2.35M 🧍 human cCREs
- 927k 🐭 mouse cCREs
www.biorxiv.org/content/10.1...
Led by @moorejille.bsky.social, this preprint summarizes data and analyses generated by hundreds of contributors across ENCODE 4
The ENCODE 4 expanded registry of regulatory elements
- 2.35M 🧍 human cCREs
- 927k 🐭 mouse cCREs
www.biorxiv.org/content/10.1...
Led by @moorejille.bsky.social, this preprint summarizes data and analyses generated by hundreds of contributors across ENCODE 4
January 8, 2025 at 8:35 PM
Now available as preprint:
The ENCODE 4 expanded registry of regulatory elements
- 2.35M 🧍 human cCREs
- 927k 🐭 mouse cCREs
www.biorxiv.org/content/10.1...
Led by @moorejille.bsky.social, this preprint summarizes data and analyses generated by hundreds of contributors across ENCODE 4
The ENCODE 4 expanded registry of regulatory elements
- 2.35M 🧍 human cCREs
- 927k 🐭 mouse cCREs
www.biorxiv.org/content/10.1...
Led by @moorejille.bsky.social, this preprint summarizes data and analyses generated by hundreds of contributors across ENCODE 4
Reposted by Tijs van Lieshout

Super excited to announce our latest flagship model Borzoi: major props to Johannes & David Kelley et al for advancing it. It's been a long journey from our prior Enformer model into this one. A few innovations: i) longer DNA context, ii) adaptation to predict RNA-seq abundance and splice isoforms,
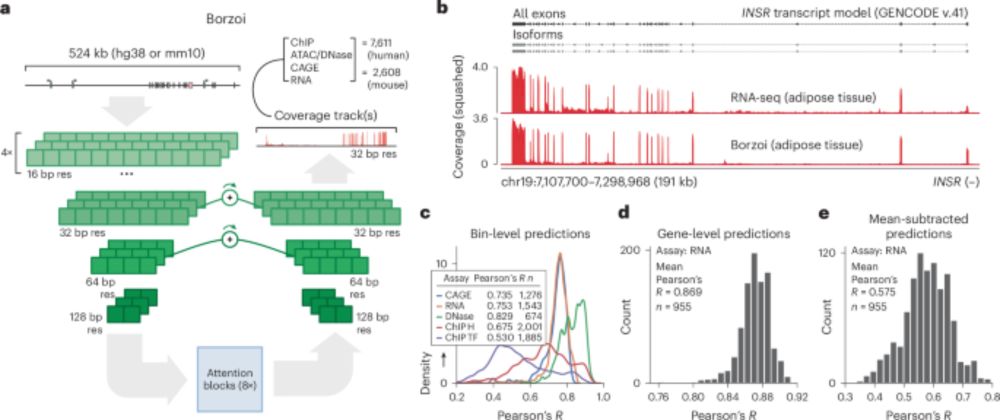
Predicting RNA-seq coverage from DNA sequence as a unifying model of gene regulation - Nature Genetics
Borzoi adapts the Enformer sequence-to-expression model to directly predict RNA-seq coverage, enabling the in-silico analysis of variant effects across multiple layers of gene regulation.
www.nature.com
January 9, 2025 at 3:08 AM
Super excited to announce our latest flagship model Borzoi: major props to Johannes & David Kelley et al for advancing it. It's been a long journey from our prior Enformer model into this one. A few innovations: i) longer DNA context, ii) adaptation to predict RNA-seq abundance and splice isoforms,
Reposted by Tijs van Lieshout
Our ChromBPNet preprint out!
www.biorxiv.org/content/10.1...
Huge congrats to Anusri! This was quite a slog (for both of us) but we r very proud of this one! It is a long read but worth it IMHO. Methods r in the supp. materials. Bluetorial coming soon below 1/
www.biorxiv.org/content/10.1...
Huge congrats to Anusri! This was quite a slog (for both of us) but we r very proud of this one! It is a long read but worth it IMHO. Methods r in the supp. materials. Bluetorial coming soon below 1/
December 25, 2024 at 11:48 PM
Our ChromBPNet preprint out!
www.biorxiv.org/content/10.1...
Huge congrats to Anusri! This was quite a slog (for both of us) but we r very proud of this one! It is a long read but worth it IMHO. Methods r in the supp. materials. Bluetorial coming soon below 1/
www.biorxiv.org/content/10.1...
Huge congrats to Anusri! This was quite a slog (for both of us) but we r very proud of this one! It is a long read but worth it IMHO. Methods r in the supp. materials. Bluetorial coming soon below 1/
Reposted by Tijs van Lieshout

A month ago we @vevotherapeutics.bsky.social announced that we have generated the largest single-cell perturbation atlas in history, Tahoe-100M. Today, we announce that we will fully open-source Tahoe-100M in Feb, as part of a collaboration with NVidia health to train cell state models.
January 13, 2025 at 4:23 PM
A month ago we @vevotherapeutics.bsky.social announced that we have generated the largest single-cell perturbation atlas in history, Tahoe-100M. Today, we announce that we will fully open-source Tahoe-100M in Feb, as part of a collaboration with NVidia health to train cell state models.
Reposted by Tijs van Lieshout

You have to believe you can write a manuscript in a week, so that it will end up only taking three months.
December 10, 2024 at 5:00 PM
You have to believe you can write a manuscript in a week, so that it will end up only taking three months.
Reposted by Tijs van Lieshout

Hey, a question for the genetics community. Does genetic fine-mapping work well? How often does it miss?
We usually find that most fine-mapped variants do not fall within coding or regulatory regions. Is it a limitation of epigenomics or a limitation of fine-mapping? Please share your thoughts!
We usually find that most fine-mapped variants do not fall within coding or regulatory regions. Is it a limitation of epigenomics or a limitation of fine-mapping? Please share your thoughts!
November 19, 2024 at 7:56 PM
Hey, a question for the genetics community. Does genetic fine-mapping work well? How often does it miss?
We usually find that most fine-mapped variants do not fall within coding or regulatory regions. Is it a limitation of epigenomics or a limitation of fine-mapping? Please share your thoughts!
We usually find that most fine-mapped variants do not fall within coding or regulatory regions. Is it a limitation of epigenomics or a limitation of fine-mapping? Please share your thoughts!
Reposted by Tijs van Lieshout

We have a method for fast GWAS data access. STABIX adds a p-value index that with a position index (what TABIX has) allows you to quickly retrieve, eg. all significant SNPs in a target gene. Also column compression to reduce the size. www.biorxiv.org/content/10.1...
github.com/kristen-schn...
github.com/kristen-schn...

STABIX: Summary statistic-based GWAS indexing and compression
Genome-Wide Association Studies (GWAS) are widely used to investigate the role of genetics in disease traits, but the resulting file sizes from these studies are large, posing barriers to efficient st...
www.biorxiv.org
November 21, 2024 at 3:11 PM
We have a method for fast GWAS data access. STABIX adds a p-value index that with a position index (what TABIX has) allows you to quickly retrieve, eg. all significant SNPs in a target gene. Also column compression to reduce the size. www.biorxiv.org/content/10.1...
github.com/kristen-schn...
github.com/kristen-schn...
Reposted by Tijs van Lieshout
Saying ‘I don’t know’ in talks has become a lost art, unfortunately. Super interesting discussions require us to be open about what we don’t know and what still needs to be explored. Here’s my recipe for answering questions in talks:

November 23, 2024 at 8:41 PM
Saying ‘I don’t know’ in talks has become a lost art, unfortunately. Super interesting discussions require us to be open about what we don’t know and what still needs to be explored. Here’s my recipe for answering questions in talks:
Reposted by Tijs van Lieshout

Looking for Adobe alternatives but don't really know where to look?
This list is pretty dang awesome: github.com/KenneyNL/Ado...
#gamedev
This list is pretty dang awesome: github.com/KenneyNL/Ado...
#gamedev

GitHub - KenneyNL/Adobe-Alternatives: A list of alternatives for Adobe software
A list of alternatives for Adobe software. Contribute to KenneyNL/Adobe-Alternatives development by creating an account on GitHub.
github.com
November 24, 2024 at 2:33 PM
Looking for Adobe alternatives but don't really know where to look?
This list is pretty dang awesome: github.com/KenneyNL/Ado...
#gamedev
This list is pretty dang awesome: github.com/KenneyNL/Ado...
#gamedev
Reposted by Tijs van Lieshout

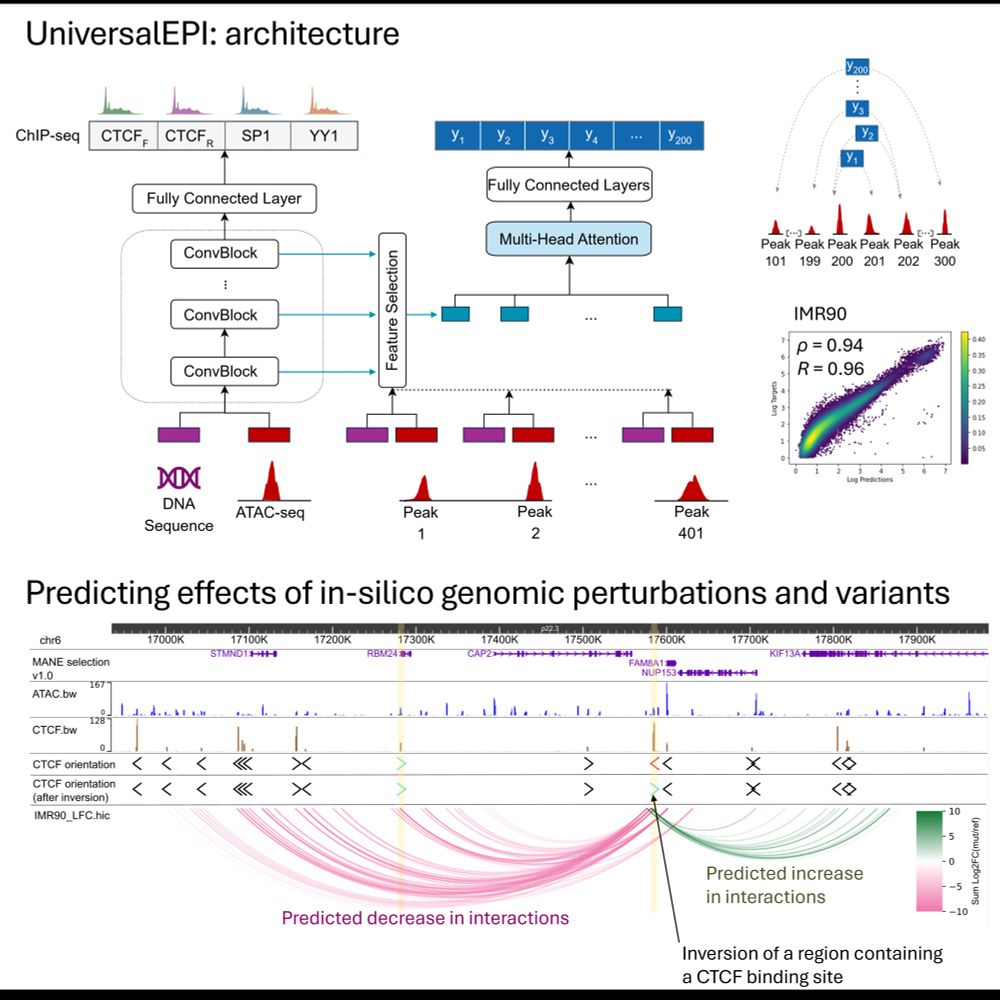
🚀 New preprint from our lab, Ekaterina Krymova, and @fabiantheis.bsky.social: UniversalEPI, an attention-based method to predict enhancer-promoter interactions from DNA sequence and ATAC-seq🌟 Read the full preprint: www.biorxiv.org/content/10.1... by @aayushgrover.bsky.social, L. Zhang & I.L. Ibarra
November 26, 2024 at 1:40 PM
🚀 New preprint from our lab, Ekaterina Krymova, and @fabiantheis.bsky.social: UniversalEPI, an attention-based method to predict enhancer-promoter interactions from DNA sequence and ATAC-seq🌟 Read the full preprint: www.biorxiv.org/content/10.1... by @aayushgrover.bsky.social, L. Zhang & I.L. Ibarra
Reposted by Tijs van Lieshout
What a surprise (not!). Yet again ... poor evaluations of specialized medical LLMs result in overhyped claims relative to the base LLMs. #bioMLeval

Medically adapted foundation models (think Med-*) turn out to be more hot air than hot stuff. Correcting for fatal flaws in evaluation, the current crop are no better on balance than generic foundation models, even on the very tasks for which benefits are claimed.
arxiv.org/abs/2411.04118
arxiv.org/abs/2411.04118

Medical Adaptation of Large Language and Vision-Language Models: Are We Making Progress?
Several recent works seek to develop foundation models specifically for medical applications, adapting general-purpose large language models (LLMs) and vision-language models (VLMs) via continued pret...
arxiv.org
November 27, 2024 at 2:16 AM
What a surprise (not!). Yet again ... poor evaluations of specialized medical LLMs result in overhyped claims relative to the base LLMs. #bioMLeval
Reposted by Tijs van Lieshout
Gene regulation involves thousands of proteins that bind DNA, yet comprehensively mapping these is challenging. Our paper in Nature Genetics describes ChIP-DIP, a method for genome-wide mapping of hundreds of DNA-protein interactions in a single experiment.
www.nature.com/articles/s41...
www.nature.com/articles/s41...
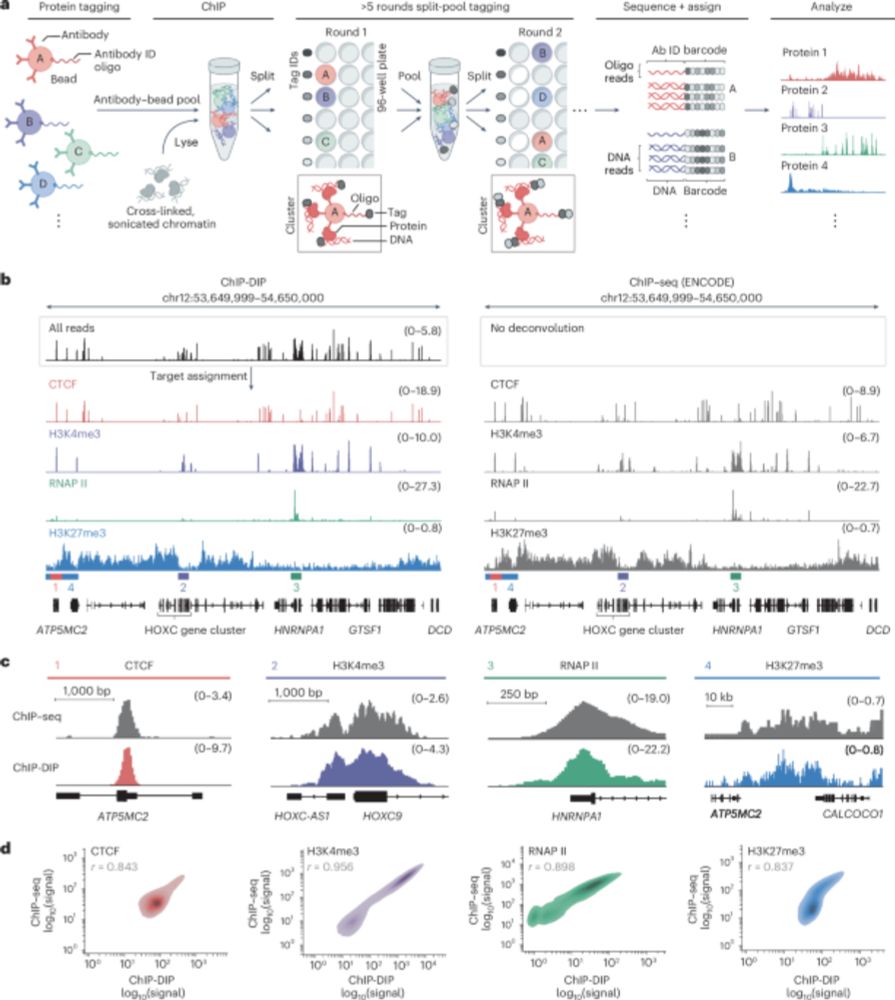
ChIP-DIP maps binding of hundreds of proteins to DNA simultaneously and identifies diverse gene regulatory elements - Nature Genetics
ChIP-DIP (ChIP done in parallel) is a highly multiplex assay for protein–DNA binding, scalable to hundreds of proteins including modified histones, chromatin regulators and transcription factors, offe...
www.nature.com
November 27, 2024 at 4:13 AM
Gene regulation involves thousands of proteins that bind DNA, yet comprehensively mapping these is challenging. Our paper in Nature Genetics describes ChIP-DIP, a method for genome-wide mapping of hundreds of DNA-protein interactions in a single experiment.
www.nature.com/articles/s41...
www.nature.com/articles/s41...