


juhyunk.bsky.social
@juhyunk.bsky.social
Reposted by juhyunk.bsky.social
Now published in GigaScience with minor improvements: academic.oup.com/gigascience/...
* Download: zenodo.org/records/1490...
* More info: github.com/lh3/panmask
* Download: zenodo.org/records/1490...
* More info: github.com/lh3/panmask
Preprint on "Finding easy regions for short-read variant calling from pangenome data": arxiv.org/abs/2507.03718

September 4, 2025 at 4:44 PM
Now published in GigaScience with minor improvements: academic.oup.com/gigascience/...
* Download: zenodo.org/records/1490...
* More info: github.com/lh3/panmask
* Download: zenodo.org/records/1490...
* More info: github.com/lh3/panmask
Reposted by juhyunk.bsky.social

Activity of most genes is controlled by multiple enhancers, but is there activation coordinated? We leveraged Nanopore to identify a specific set of elements that are simultaneously accessible on the same DNA molecules and are coordinated in their activation. www.biorxiv.org/content/10.1...

August 18, 2025 at 12:23 PM
Activity of most genes is controlled by multiple enhancers, but is there activation coordinated? We leveraged Nanopore to identify a specific set of elements that are simultaneously accessible on the same DNA molecules and are coordinated in their activation. www.biorxiv.org/content/10.1...
Reposted by juhyunk.bsky.social

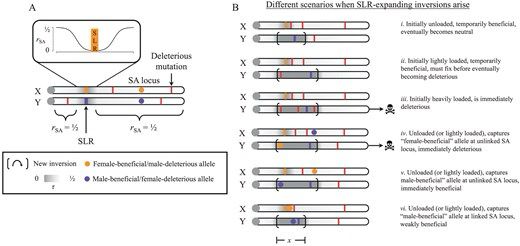
Reposted by juhyunk.bsky.social
If you want to check if a human gene has copy-number changes or lands in a complex region, try pangene.bioinweb.org. Recently updated with more and better assemblies.

April 26, 2025 at 1:06 AM
If you want to check if a human gene has copy-number changes or lands in a complex region, try pangene.bioinweb.org. Recently updated with more and better assemblies.
Reposted by juhyunk.bsky.social

AlphaGenome deepmind.google/discover/blo... 🧬🖥️🧪
API: github.com/google-deepm...
Docs: www.alphagenomedocs.com
Community: www.alphagenomecommunity.com
API: github.com/google-deepm...
Docs: www.alphagenomedocs.com
Community: www.alphagenomecommunity.com
June 25, 2025 at 4:53 PM
AlphaGenome deepmind.google/discover/blo... 🧬🖥️🧪
API: github.com/google-deepm...
Docs: www.alphagenomedocs.com
Community: www.alphagenomecommunity.com
API: github.com/google-deepm...
Docs: www.alphagenomedocs.com
Community: www.alphagenomecommunity.com
Reposted by juhyunk.bsky.social
Improving gene isoform quantification with miniQuant www.nature.com/articles/s41... 🧬🖥️🧪 github.com/Augroup/mini... (no code, only executables, noncommercial license)


June 20, 2025 at 3:31 PM
Improving gene isoform quantification with miniQuant www.nature.com/articles/s41... 🧬🖥️🧪 github.com/Augroup/mini... (no code, only executables, noncommercial license)
Reposted by juhyunk.bsky.social

Congrats to @dantipov.bsky.social et al. on the publication of Verkko2! The team put a ton of work into this making it the first assembler that deals with the complexity of human acrocentric chromosomes. Lots of interesting discoveries to come! genome.cshlp.org/content/earl...

June 17, 2025 at 1:39 PM
Congrats to @dantipov.bsky.social et al. on the publication of Verkko2! The team put a ton of work into this making it the first assembler that deals with the complexity of human acrocentric chromosomes. Lots of interesting discoveries to come! genome.cshlp.org/content/earl...
Reposted by juhyunk.bsky.social

Now published! Note that since Vikram's original post (quoted here), he's made it easy to dynamically update a set of multi-MUMs (e.g. when more genomes are added to a pangenome) and to find multi-MUMs for huge collections like HPRCv2 genomebiology.biomedcentral.com/articles/10....
June 17, 2025 at 2:02 PM
Now published! Note that since Vikram's original post (quoted here), he's made it easy to dynamically update a set of multi-MUMs (e.g. when more genomes are added to a pangenome) and to find multi-MUMs for huge collections like HPRCv2 genomebiology.biomedcentral.com/articles/10....
Reposted by juhyunk.bsky.social

Our new contribution to the quest to find causal GWAS genes! Sam Ghatan from my lab at @nygenome.org led a systematic comparison of eQTLs and CRISPRi+scRNA-seq screens. TL;DR: they provide highly complementary insights, with ortogonal pros and cons. 🧵👇
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
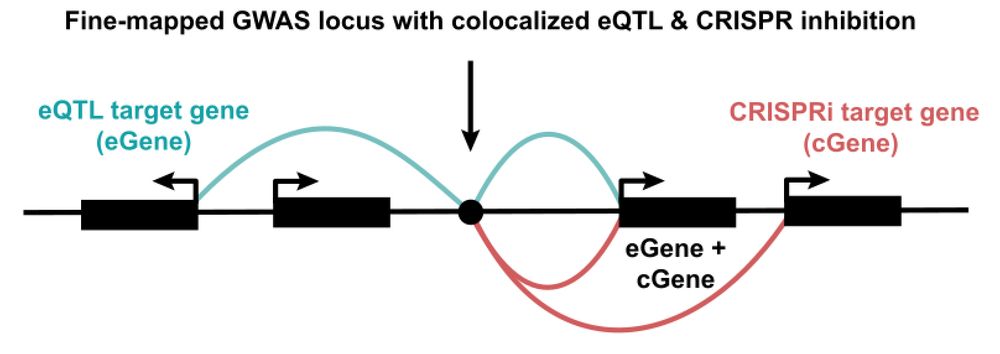
May 6, 2025 at 5:01 PM
Our new contribution to the quest to find causal GWAS genes! Sam Ghatan from my lab at @nygenome.org led a systematic comparison of eQTLs and CRISPRi+scRNA-seq screens. TL;DR: they provide highly complementary insights, with ortogonal pros and cons. 🧵👇
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
Reposted by juhyunk.bsky.social

Our featured article: DNA methylation in mammalian development and disease go.nature.com/3X14Ali #Review by Zachary D. Smith @yaleschoolofmed.bsky.social, Sara Hetzel & Alexander Meissner @molgen.mpg.de
Free to read here: rdcu.be/dQEND
Free to read here: rdcu.be/dQEND

DNA methylation in mammalian development and disease - Nature Reviews Genetics
In this Review, Smith et al. describe DNA methylation landscapes that emerge over mammalian development and within key disease states, as well as how different methyltransferases interface w...
go.nature.com
December 13, 2024 at 12:57 PM
Our featured article: DNA methylation in mammalian development and disease go.nature.com/3X14Ali #Review by Zachary D. Smith @yaleschoolofmed.bsky.social, Sara Hetzel & Alexander Meissner @molgen.mpg.de
Free to read here: rdcu.be/dQEND
Free to read here: rdcu.be/dQEND
Reposted by juhyunk.bsky.social

A tool to dissect regulatory DNA in its endogenous context enables the identification of designed small edits (<10 bp) that fine-tune gene expression, with potential therapeutic implications #NBThighlight www.cell.com/cell/abstrac...
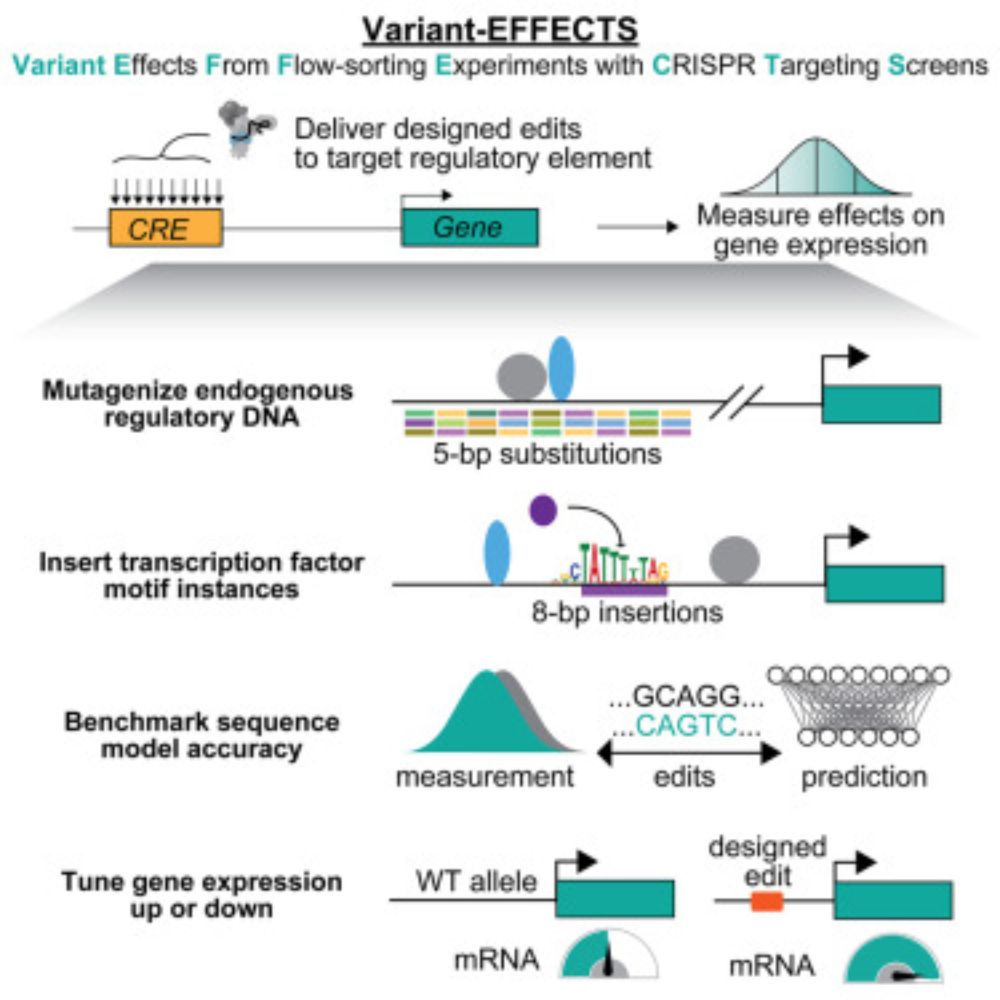
Rewriting regulatory DNA to dissect and reprogram gene expression
Martyn and Montgomery et al. present Variant-EFFECTS, a high-throughput technology
developed to precisely measure the effects of CRISPR-mediated edits on gene expression.
Variant-EFFECTS is reproducib...
www.cell.com
April 17, 2025 at 1:38 PM
A tool to dissect regulatory DNA in its endogenous context enables the identification of designed small edits (<10 bp) that fine-tune gene expression, with potential therapeutic implications #NBThighlight www.cell.com/cell/abstrac...
Reposted by juhyunk.bsky.social

Evolutionary divergence in CTCF-mediated chromatin topology drives transcriptional innovation in humans www.nature.com/articles/s41...
Human vs macaque:
▶️2133 gained (2.7%), 2418 lost (3.1%) CTCF loops in B-cells
▶️5873 gained (7.3%), 6708 lost (8.3%) in neurons
▶️implications for ASD and immunity
Human vs macaque:
▶️2133 gained (2.7%), 2418 lost (3.1%) CTCF loops in B-cells
▶️5873 gained (7.3%), 6708 lost (8.3%) in neurons
▶️implications for ASD and immunity

March 30, 2025 at 2:19 PM
Evolutionary divergence in CTCF-mediated chromatin topology drives transcriptional innovation in humans www.nature.com/articles/s41...
Human vs macaque:
▶️2133 gained (2.7%), 2418 lost (3.1%) CTCF loops in B-cells
▶️5873 gained (7.3%), 6708 lost (8.3%) in neurons
▶️implications for ASD and immunity
Human vs macaque:
▶️2133 gained (2.7%), 2418 lost (3.1%) CTCF loops in B-cells
▶️5873 gained (7.3%), 6708 lost (8.3%) in neurons
▶️implications for ASD and immunity
Reposted by juhyunk.bsky.social

Interested in Comparative Genomics?
Join us to learn genome assemby&annotation, variant detection, and evolutionary analysis with lots of hands-on sessions! @nanoporetech.com @pacbio.bsky.social @sedlazeck.bsky.social
www.physalia-courses.org/courses-work...
Join us to learn genome assemby&annotation, variant detection, and evolutionary analysis with lots of hands-on sessions! @nanoporetech.com @pacbio.bsky.social @sedlazeck.bsky.social
www.physalia-courses.org/courses-work...

March 27, 2025 at 3:40 PM
Interested in Comparative Genomics?
Join us to learn genome assemby&annotation, variant detection, and evolutionary analysis with lots of hands-on sessions! @nanoporetech.com @pacbio.bsky.social @sedlazeck.bsky.social
www.physalia-courses.org/courses-work...
Join us to learn genome assemby&annotation, variant detection, and evolutionary analysis with lots of hands-on sessions! @nanoporetech.com @pacbio.bsky.social @sedlazeck.bsky.social
www.physalia-courses.org/courses-work...
Reposted by juhyunk.bsky.social

Newly published in Genetics:
academic.oup.com/genetics/adv...
We ask what gene flow & introgression do to gene coexpression networks. The answer is: a lot.
academic.oup.com/genetics/adv...
We ask what gene flow & introgression do to gene coexpression networks. The answer is: a lot.
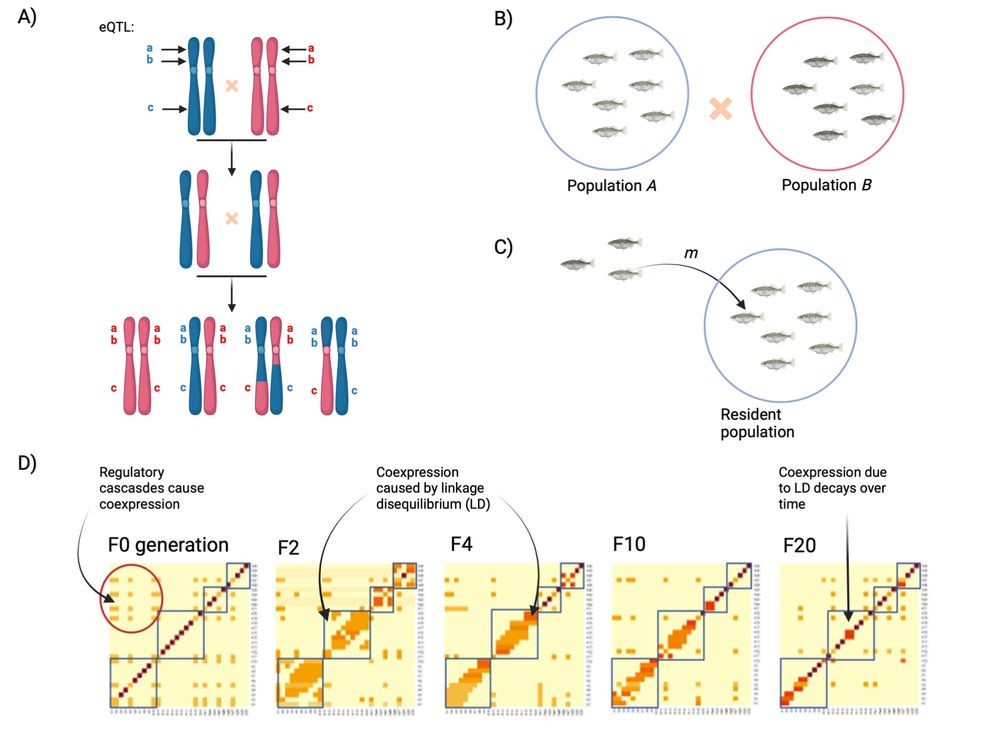
March 27, 2025 at 6:13 PM
Newly published in Genetics:
academic.oup.com/genetics/adv...
We ask what gene flow & introgression do to gene coexpression networks. The answer is: a lot.
academic.oup.com/genetics/adv...
We ask what gene flow & introgression do to gene coexpression networks. The answer is: a lot.
Reposted by juhyunk.bsky.social

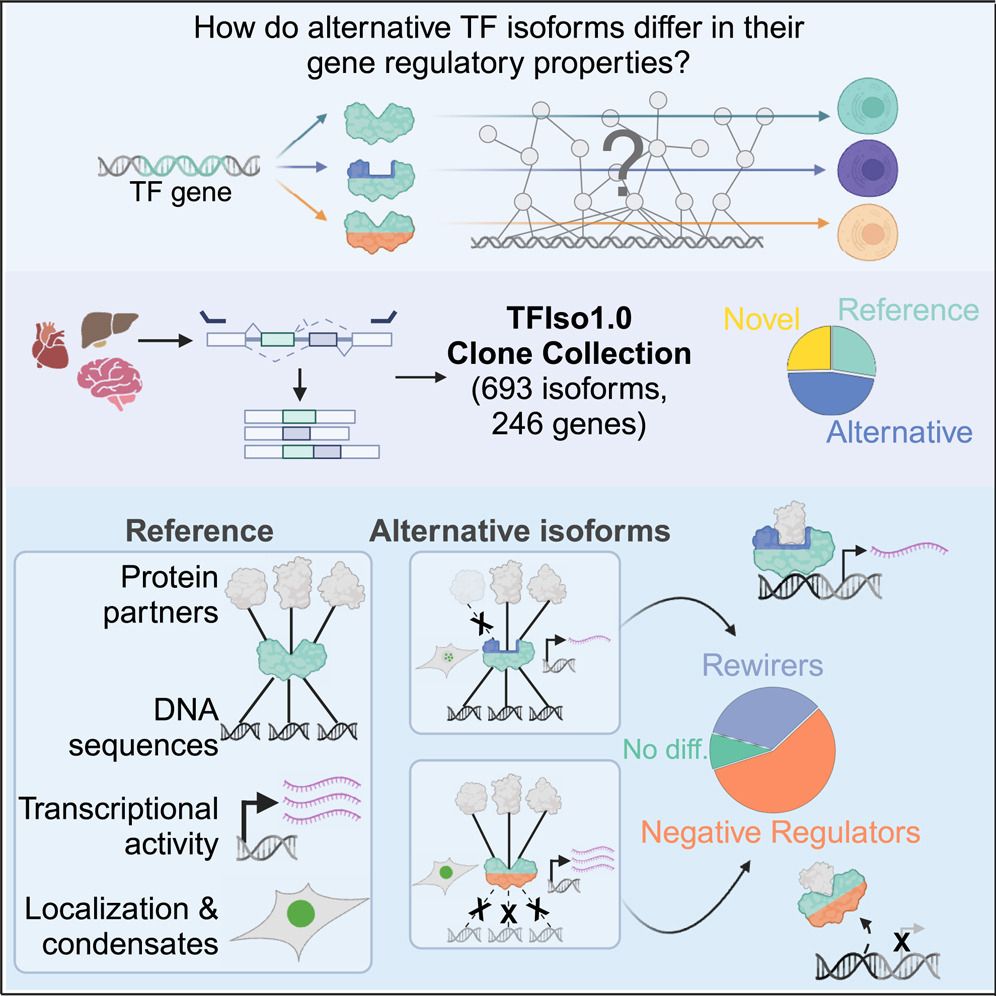
our work on the molecular differences between transcription factor isoforms is out now in Molecular Cell!
key point: 2/3rds of TF isos differ in properties like DNA binding & transcriptional activity
many are "negative regulators" & misexpressed in cancer
www.sciencedirect.com/science/arti...
key point: 2/3rds of TF isos differ in properties like DNA binding & transcriptional activity
many are "negative regulators" & misexpressed in cancer
www.sciencedirect.com/science/arti...
March 26, 2025 at 5:11 PM
our work on the molecular differences between transcription factor isoforms is out now in Molecular Cell!
key point: 2/3rds of TF isos differ in properties like DNA binding & transcriptional activity
many are "negative regulators" & misexpressed in cancer
www.sciencedirect.com/science/arti...
key point: 2/3rds of TF isos differ in properties like DNA binding & transcriptional activity
many are "negative regulators" & misexpressed in cancer
www.sciencedirect.com/science/arti...
Reposted by juhyunk.bsky.social

A new article from Laurel Hiatt and @hdashnow.bsky.social describing STRchive is now available at Genome Medicine. genomemedicine.biomedcentral.com/articles/10....
Check out the database resource, as STRchive "streamlines TR variant interpretation at disease-associated loci."
strchive.org
Check out the database resource, as STRchive "streamlines TR variant interpretation at disease-associated loci."
strchive.org
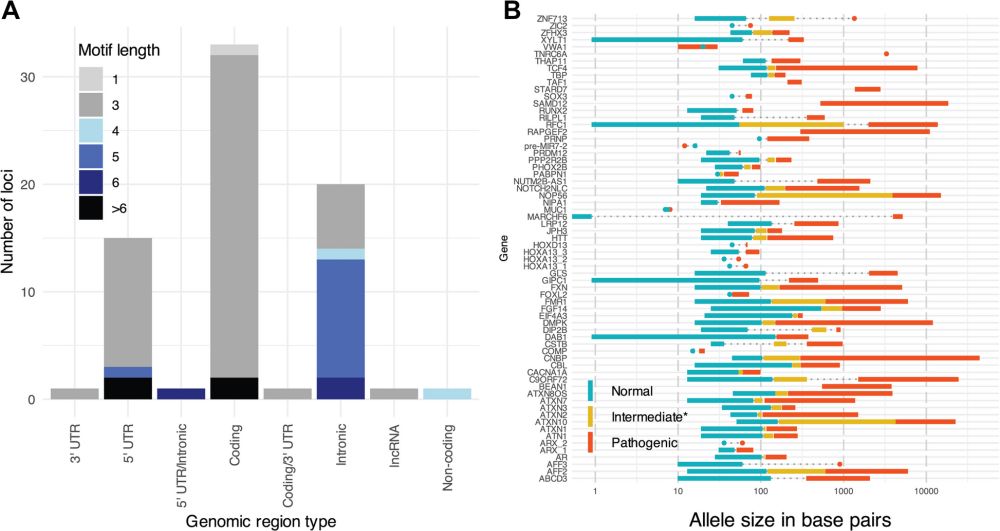
STRchive: a dynamic resource detailing population-level and locus-specific insights at tandem repeat disease loci - Genome Medicine
Approximately 8% of the human genome consists of repetitive elements called tandem repeats (TRs): short tandem repeats (STRs) of 1–6 bp motifs and variable number tandem repeats (VNTRs) of 7 + bp moti...
genomemedicine.biomedcentral.com
March 26, 2025 at 7:18 PM
A new article from Laurel Hiatt and @hdashnow.bsky.social describing STRchive is now available at Genome Medicine. genomemedicine.biomedcentral.com/articles/10....
Check out the database resource, as STRchive "streamlines TR variant interpretation at disease-associated loci."
strchive.org
Check out the database resource, as STRchive "streamlines TR variant interpretation at disease-associated loci."
strchive.org
Reposted by juhyunk.bsky.social
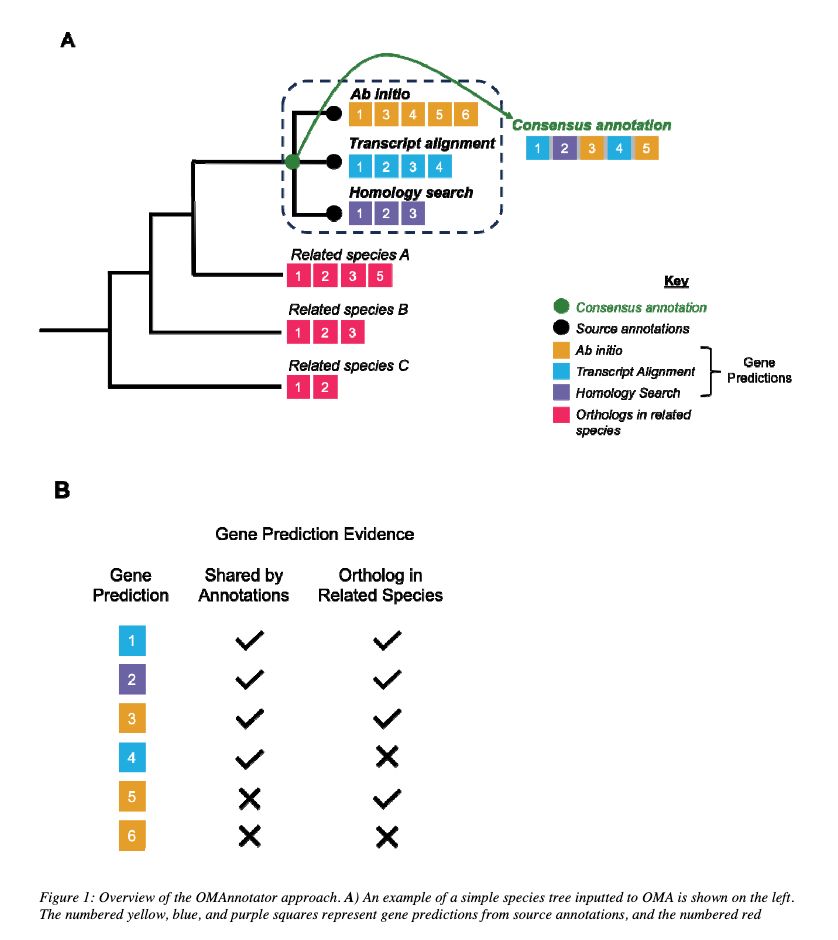
OMAnnotator: a novel approach to building an annotated consensus genome sequence www.biorxiv.org/content/10.1... 🧬🖥️🧪 github.com/DessimozLab/...



March 13, 2025 at 3:30 PM
OMAnnotator: a novel approach to building an annotated consensus genome sequence www.biorxiv.org/content/10.1... 🧬🖥️🧪 github.com/DessimozLab/...
Reposted by juhyunk.bsky.social

🧪 The MorPhIC Consortium is building a comprehensive catalog of human gene functions and their roles in disease using in vitro multicellular systems. Discover more about their ambitious goal and approach in @nature.com here: www.nature.com/articles/s41....

MorPhiC Consortium: towards functional characterization of all human genes - Nature
This Perspective discusses strategies and challenges for the Molecular Phenotypes of Null Alleles in Cells (MorPhiC) Consortium as it aims to catalogue the molecular and cellular phenotypes associated...
www.nature.com
March 6, 2025 at 12:52 PM
🧪 The MorPhIC Consortium is building a comprehensive catalog of human gene functions and their roles in disease using in vitro multicellular systems. Discover more about their ambitious goal and approach in @nature.com here: www.nature.com/articles/s41....
Reposted by juhyunk.bsky.social

New preprint is out!
We investigate how well you can call variants directly from genome assemblies compared to traditional read-based variant calling.
Read it here: www.biorxiv.org/content/10.1...
Data & code: github.com/rrwick/Are-r...
(1/8)
We investigate how well you can call variants directly from genome assemblies compared to traditional read-based variant calling.
Read it here: www.biorxiv.org/content/10.1...
Data & code: github.com/rrwick/Are-r...
(1/8)

Are reads required? High-precision variant calling from bacterial genome assemblies
Accurate nucleotide variant calling is essential in microbial genomics, particularly for outbreak tracking and phylogenetics. This study evaluates variant calls derived from genome assemblies compared...
www.biorxiv.org
March 3, 2025 at 3:24 AM
New preprint is out!
We investigate how well you can call variants directly from genome assemblies compared to traditional read-based variant calling.
Read it here: www.biorxiv.org/content/10.1...
Data & code: github.com/rrwick/Are-r...
(1/8)
We investigate how well you can call variants directly from genome assemblies compared to traditional read-based variant calling.
Read it here: www.biorxiv.org/content/10.1...
Data & code: github.com/rrwick/Are-r...
(1/8)
Reposted by juhyunk.bsky.social
Accurate Somatic SV detection via sequence graph model-based local pan-genome optimization https://www.biorxiv.org/content/10.1101/2025.02.11.636543v1 🧬🖥️🧪 https://github.com/Goatofmountain/TDScope


February 17, 2025 at 9:00 PM
Accurate Somatic SV detection via sequence graph model-based local pan-genome optimization https://www.biorxiv.org/content/10.1101/2025.02.11.636543v1 🧬🖥️🧪 https://github.com/Goatofmountain/TDScope
Reposted by juhyunk.bsky.social
Comparative population pangenomes reveal unexpected complexity and fitness effects of structural variants https://www.biorxiv.org/content/10.1101/2025.02.11.637762v1 🧬🖥️🧪 https://github.com/harvardinformatics/scrub-jay-genomics


February 14, 2025 at 4:30 PM
Comparative population pangenomes reveal unexpected complexity and fitness effects of structural variants https://www.biorxiv.org/content/10.1101/2025.02.11.637762v1 🧬🖥️🧪 https://github.com/harvardinformatics/scrub-jay-genomics
Reposted by juhyunk.bsky.social

We're delighted to share our work on scrambling the human genome using prime editing, repetitive elements, and recombinases in @science.org , led by @jonaskoeppel.bsky.social , @f-raphael.bsky.social , with @proftomellis.bsky.social and George Church.
www.science.org/doi/10.1126/...
www.science.org/doi/10.1126/...
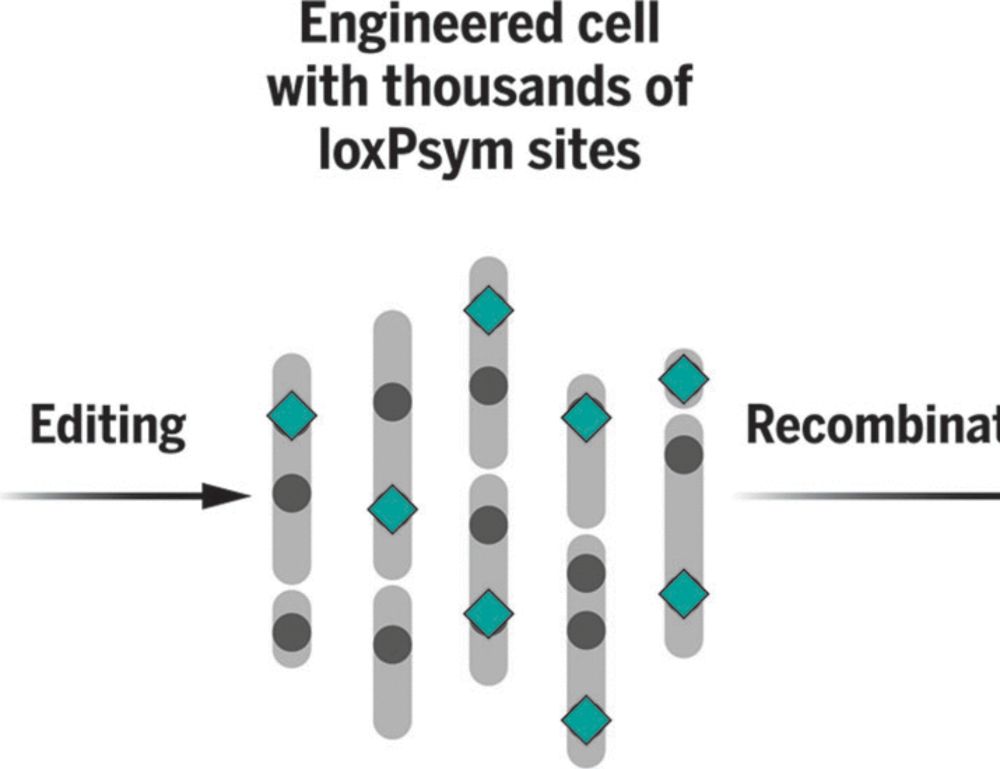
Randomizing the human genome by engineering recombination between repeat elements
We lack tools to edit DNA sequences at scales necessary to study 99% of the human genome that is noncoding. To address this gap, we applied CRISPR prime editing to insert recombination handles into re...
www.science.org
January 31, 2025 at 1:49 PM
We're delighted to share our work on scrambling the human genome using prime editing, repetitive elements, and recombinases in @science.org , led by @jonaskoeppel.bsky.social , @f-raphael.bsky.social , with @proftomellis.bsky.social and George Church.
www.science.org/doi/10.1126/...
www.science.org/doi/10.1126/...
Reposted by juhyunk.bsky.social
This project stands together with work from @sudpinglay.bsky.social and @jshendure.bsky.social , who came up with an alternative method that includes a clever, single-cell sequencing strategy to read out the variants generated by scrambling.
www.science.org/doi/10.1126/...
www.science.org/doi/10.1126/...
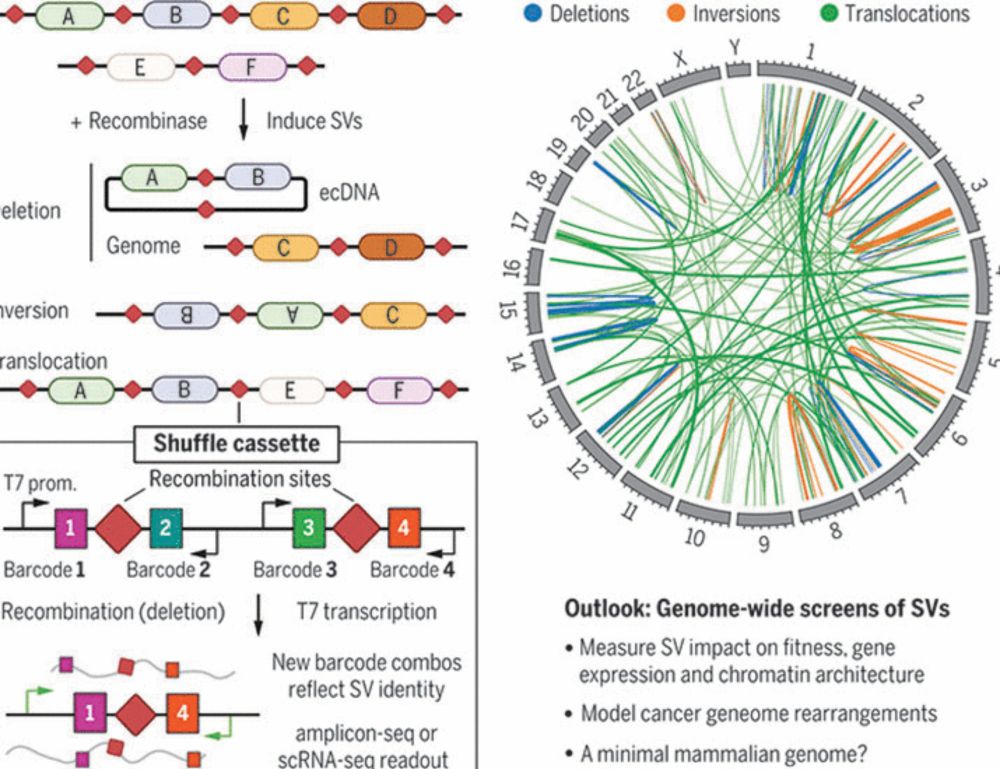
Multiplex generation and single-cell analysis of structural variants in mammalian genomes
Studying the functional consequences of structural variants (SVs) in mammalian genomes is challenging because (i) SVs arise much less commonly than single-nucleotide variants or small indels and (ii) ...
www.science.org
January 31, 2025 at 1:49 PM
This project stands together with work from @sudpinglay.bsky.social and @jshendure.bsky.social , who came up with an alternative method that includes a clever, single-cell sequencing strategy to read out the variants generated by scrambling.
www.science.org/doi/10.1126/...
www.science.org/doi/10.1126/...
Reposted by juhyunk.bsky.social

This is very interesting and highly relevant paper to clinical genetics and rare disease researchers. I wrote the accompanying News & Views piece. Free sharing link here: rdcu.be/d0VlK
November 20, 2024 at 11:11 PM
This is very interesting and highly relevant paper to clinical genetics and rare disease researchers. I wrote the accompanying News & Views piece. Free sharing link here: rdcu.be/d0VlK