


Jim Shaw
@jimshaw.bsky.social
Postdoc at Dana-Farber and Harvard Med with Heng Li (@lh3lh3.bsky.social). Prev: UBC / UofT.
I like thinking about computational biological sequence analysis and its applications to metagenomics.
https://jim-shaw-bluenote.github.io
I like thinking about computational biological sequence analysis and its applications to metagenomics.
https://jim-shaw-bluenote.github.io
Reposted by Jim Shaw

🚨New preprint out!
We present a foundational genomic resource of human gut microbiome viruses. It delivers high-quality, deeply curated data spanning taxonomy, predicted hosts, structures, and functions, providing a reference for gut virome research. (1/8)
www.biorxiv.org/content/10.1...
We present a foundational genomic resource of human gut microbiome viruses. It delivers high-quality, deeply curated data spanning taxonomy, predicted hosts, structures, and functions, providing a reference for gut virome research. (1/8)
www.biorxiv.org/content/10.1...

November 6, 2025 at 5:26 PM
🚨New preprint out!
We present a foundational genomic resource of human gut microbiome viruses. It delivers high-quality, deeply curated data spanning taxonomy, predicted hosts, structures, and functions, providing a reference for gut virome research. (1/8)
www.biorxiv.org/content/10.1...
We present a foundational genomic resource of human gut microbiome viruses. It delivers high-quality, deeply curated data spanning taxonomy, predicted hosts, structures, and functions, providing a reference for gut virome research. (1/8)
www.biorxiv.org/content/10.1...
Reposted by Jim Shaw

Excited to share our LongTrack study out in
@natmicrobiol.nature.com today!
Fecal microbiota transplant (FMT), donor 💩 => patients' gut, is an effective treatment for recurrent C. difficile infection & is being evaluated for Inflammatory Bowel Diseases (IBD) & other conditions 1/
📄 rdcu.be/eL8mR
@natmicrobiol.nature.com today!
Fecal microbiota transplant (FMT), donor 💩 => patients' gut, is an effective treatment for recurrent C. difficile infection & is being evaluated for Inflammatory Bowel Diseases (IBD) & other conditions 1/
📄 rdcu.be/eL8mR

Long-read metagenomics for strain tracking after faecal microbiota transplant
Nature Microbiology - A long-read metagenomics method empowers faecal microbiota transplantation studies by precisely tracking bacteria from donors to recipients, distinguishing co-existing strains...
rdcu.be
October 22, 2025 at 3:39 PM
Excited to share our LongTrack study out in
@natmicrobiol.nature.com today!
Fecal microbiota transplant (FMT), donor 💩 => patients' gut, is an effective treatment for recurrent C. difficile infection & is being evaluated for Inflammatory Bowel Diseases (IBD) & other conditions 1/
📄 rdcu.be/eL8mR
@natmicrobiol.nature.com today!
Fecal microbiota transplant (FMT), donor 💩 => patients' gut, is an effective treatment for recurrent C. difficile infection & is being evaluated for Inflammatory Bowel Diseases (IBD) & other conditions 1/
📄 rdcu.be/eL8mR
Reposted by Jim Shaw

Our @narjournal.bsky.social manuscript is out! It explores the growth of the GTDB (gtdb.ecogenomic.org) since its inception, as well as updates to the website, methodology, policies, and major taxonomic and nomenclatural changes over the past three years.
academic.oup.com/nar/advance-...
academic.oup.com/nar/advance-...

GTDB release 10: a complete and systematic taxonomy for 715 230 bacterial and 17 245 archaeal genomes
Abstract. The Genome Taxonomy Database (GTDB; https://gtdb.ecogenomic.org) provides a phylogenetically consistent and rank normalized genome-based taxonomy
academic.oup.com
October 22, 2025 at 2:20 PM
Our @narjournal.bsky.social manuscript is out! It explores the growth of the GTDB (gtdb.ecogenomic.org) since its inception, as well as updates to the website, methodology, policies, and major taxonomic and nomenclatural changes over the past three years.
academic.oup.com/nar/advance-...
academic.oup.com/nar/advance-...
Reposted by Jim Shaw

Our preprint on our new metagenomic HiFi assembler Alice is out 🥳 Based on a *new sketching method* (🧵1/6)
👉 Preprint www.biorxiv.org/content/10.1...
👉 Github github.com/rolandfaure/...
👉 Preprint www.biorxiv.org/content/10.1...
👉 Github github.com/rolandfaure/...

Alice: fast and haplotype-aware assembly of high-fidelity reads based on MSR sketching
We introduce Mapping-friendly Sequence Reduction (MSR) sketches, a sketching method for high-fidelity (HiFi) long reads, and Alice, an assembler that operates directly on these sketches. MSR produces ...
www.biorxiv.org
October 3, 2025 at 2:51 PM
Our preprint on our new metagenomic HiFi assembler Alice is out 🥳 Based on a *new sketching method* (🧵1/6)
👉 Preprint www.biorxiv.org/content/10.1...
👉 Github github.com/rolandfaure/...
👉 Preprint www.biorxiv.org/content/10.1...
👉 Github github.com/rolandfaure/...
Reposted by Jim Shaw

New pre-print from the Banfield lab, highlighting an interesting case of 1.5Mb megaplasmids found in human gut.
Plasmid genomes were resolved using #PacBio HiFi sequencing with hifiasm-meta for #metagenome assembly. Host association was detected using epigenetic signals.
doi.org/10.1101/2025...
Plasmid genomes were resolved using #PacBio HiFi sequencing with hifiasm-meta for #metagenome assembly. Host association was detected using epigenetic signals.
doi.org/10.1101/2025...

Megaplasmids associate with Escherichia coli and other Enterobacteriaceae
Humans and animals are ubiquitously colonized by Enterobacteriaceae , a bacterial family that contains both commensals and clinically significant pathogens. Here, we report Enterobacteriaceae megaplas...
doi.org
October 1, 2025 at 4:44 PM
New pre-print from the Banfield lab, highlighting an interesting case of 1.5Mb megaplasmids found in human gut.
Plasmid genomes were resolved using #PacBio HiFi sequencing with hifiasm-meta for #metagenome assembly. Host association was detected using epigenetic signals.
doi.org/10.1101/2025...
Plasmid genomes were resolved using #PacBio HiFi sequencing with hifiasm-meta for #metagenome assembly. Host association was detected using epigenetic signals.
doi.org/10.1101/2025...
Reposted by Jim Shaw
Do you know ~60% of human SVs fall in ~1% of GRCh38? See our new preprint: arxiv.org/abs/2509.23057 and the companion blog post on how we started this project and longdust: lh3.github.io/2025/09/29/o.... Work with Alvin Qin

September 30, 2025 at 2:19 AM
Do you know ~60% of human SVs fall in ~1% of GRCh38? See our new preprint: arxiv.org/abs/2509.23057 and the companion blog post on how we started this project and longdust: lh3.github.io/2025/09/29/o.... Work with Alvin Qin
Reposted by Jim Shaw

High-accuracy SNV calling for bacterial isolates using deep learning with AccuSNV https://www.biorxiv.org/content/10.1101/2025.09.26.678787v1
September 29, 2025 at 6:47 PM
High-accuracy SNV calling for bacterial isolates using deep learning with AccuSNV https://www.biorxiv.org/content/10.1101/2025.09.26.678787v1
Reposted by Jim Shaw

Delighted to see our paper studying the evolution of plasmids over the last 100 years, now out! Years of work by Adrian Cazares, also Nick Thomson @sangerinstitute.bsky.social - this version much improved over the preprint. Final version should be open access, apols.
Thread 1/n
Thread 1/n

September 25, 2025 at 9:29 PM
Delighted to see our paper studying the evolution of plasmids over the last 100 years, now out! Years of work by Adrian Cazares, also Nick Thomson @sangerinstitute.bsky.social - this version much improved over the preprint. Final version should be open access, apols.
Thread 1/n
Thread 1/n
Reposted by Jim Shaw

New blog post!
metaMDBG (@gaetanbenoit.bsky.social) and Myloasm (@jimshaw.bsky.social) have had recent releases, so I updated the benchmarks from the Autocycler paper:
rrwick.github.io/2025/09/23/a...
Both tools improved considerably! Time to update your conda environments 😄
metaMDBG (@gaetanbenoit.bsky.social) and Myloasm (@jimshaw.bsky.social) have had recent releases, so I updated the benchmarks from the Autocycler paper:
rrwick.github.io/2025/09/23/a...
Both tools improved considerably! Time to update your conda environments 😄
Benchmark update: metaMDBG and Myloasm
a blog for miscellaneous bioinformatics stuff
rrwick.github.io
September 23, 2025 at 1:53 AM
New blog post!
metaMDBG (@gaetanbenoit.bsky.social) and Myloasm (@jimshaw.bsky.social) have had recent releases, so I updated the benchmarks from the Autocycler paper:
rrwick.github.io/2025/09/23/a...
Both tools improved considerably! Time to update your conda environments 😄
metaMDBG (@gaetanbenoit.bsky.social) and Myloasm (@jimshaw.bsky.social) have had recent releases, so I updated the benchmarks from the Autocycler paper:
rrwick.github.io/2025/09/23/a...
Both tools improved considerably! Time to update your conda environments 😄
Reposted by Jim Shaw

Many of the most complex and useful functions in biology emerge at the scale of whole genomes.
Today, we share our preprint “Generative design of novel bacteriophages with genome language models”, where we validate the first, functional AI-generated genomes 🧵
Today, we share our preprint “Generative design of novel bacteriophages with genome language models”, where we validate the first, functional AI-generated genomes 🧵
September 17, 2025 at 3:03 PM
Many of the most complex and useful functions in biology emerge at the scale of whole genomes.
Today, we share our preprint “Generative design of novel bacteriophages with genome language models”, where we validate the first, functional AI-generated genomes 🧵
Today, we share our preprint “Generative design of novel bacteriophages with genome language models”, where we validate the first, functional AI-generated genomes 🧵
Reposted by Jim Shaw
agtools: a software framework to manipulate assembly graphs https://www.biorxiv.org/content/10.1101/2025.09.14.676178v1
September 16, 2025 at 8:48 PM
agtools: a software framework to manipulate assembly graphs https://www.biorxiv.org/content/10.1101/2025.09.14.676178v1
Reposted by Jim Shaw

X-Mapper 🦠🧬🧪 - a sequence aligner developed for microbes, now on Bioconda! 🚀
• 11–24× fewer suboptimal alignments (same for human genome)
• 3–579× lower inconsistency
• improves on ~30% of reads aligned to non-target species
github.com/mathjeff/map...
bioconda.github.io/recipes/x-ma...
#microsky
• 11–24× fewer suboptimal alignments (same for human genome)
• 3–579× lower inconsistency
• improves on ~30% of reads aligned to non-target species
github.com/mathjeff/map...
bioconda.github.io/recipes/x-ma...
#microsky

September 15, 2025 at 2:32 AM
X-Mapper 🦠🧬🧪 - a sequence aligner developed for microbes, now on Bioconda! 🚀
• 11–24× fewer suboptimal alignments (same for human genome)
• 3–579× lower inconsistency
• improves on ~30% of reads aligned to non-target species
github.com/mathjeff/map...
bioconda.github.io/recipes/x-ma...
#microsky
• 11–24× fewer suboptimal alignments (same for human genome)
• 3–579× lower inconsistency
• improves on ~30% of reads aligned to non-target species
github.com/mathjeff/map...
bioconda.github.io/recipes/x-ma...
#microsky
Reposted by Jim Shaw
New blog post – A quick look at Roche's SBX
lh3.github.io/2025/09/11/a...
lh3.github.io/2025/09/11/a...

September 12, 2025 at 3:26 AM
New blog post – A quick look at Roche's SBX
lh3.github.io/2025/09/11/a...
lh3.github.io/2025/09/11/a...
Reposted by Jim Shaw

I sincerely appreciate the opportunity to visit @ebi.embl.org (thanks to the @embl.org Sabbatical fellowship). The guidance and support I received from Zam (@zaminiqbal.bsky.social), John (@bacpop.org) and other colleagues have been immensely valuable! You changed my career!❤️
Sometimes you meet absolutely incredible bioinfo-magicians.
It was a huge privilege when @shenwei356.bsky.social
joined our group for a year on an @embl.org sabbatical.
While here, he developed a new way of aligning to
millions of bacteria, called LexicMap 1/n
www.nature.com/articles/s41...
It was a huge privilege when @shenwei356.bsky.social
joined our group for a year on an @embl.org sabbatical.
While here, he developed a new way of aligning to
millions of bacteria, called LexicMap 1/n
www.nature.com/articles/s41...

Efficient sequence alignment against millions of prokaryotic genomes with LexicMap - Nature Biotechnology
LexicMap uses a fixed set of probes to efficiently query gene sequences for fast and low-memory alignment.
www.nature.com
September 10, 2025 at 9:56 AM
I sincerely appreciate the opportunity to visit @ebi.embl.org (thanks to the @embl.org Sabbatical fellowship). The guidance and support I received from Zam (@zaminiqbal.bsky.social), John (@bacpop.org) and other colleagues have been immensely valuable! You changed my career!❤️
Reposted by Jim Shaw
Sometimes you meet absolutely incredible bioinfo-magicians.
It was a huge privilege when @shenwei356.bsky.social
joined our group for a year on an @embl.org sabbatical.
While here, he developed a new way of aligning to
millions of bacteria, called LexicMap 1/n
www.nature.com/articles/s41...
It was a huge privilege when @shenwei356.bsky.social
joined our group for a year on an @embl.org sabbatical.
While here, he developed a new way of aligning to
millions of bacteria, called LexicMap 1/n
www.nature.com/articles/s41...
Efficient sequence alignment against millions of prokaryotic genomes with LexicMap - Nature Biotechnology
LexicMap uses a fixed set of probes to efficiently query gene sequences for fast and low-memory alignment.
www.nature.com
September 10, 2025 at 9:12 AM
Sometimes you meet absolutely incredible bioinfo-magicians.
It was a huge privilege when @shenwei356.bsky.social
joined our group for a year on an @embl.org sabbatical.
While here, he developed a new way of aligning to
millions of bacteria, called LexicMap 1/n
www.nature.com/articles/s41...
It was a huge privilege when @shenwei356.bsky.social
joined our group for a year on an @embl.org sabbatical.
While here, he developed a new way of aligning to
millions of bacteria, called LexicMap 1/n
www.nature.com/articles/s41...
Reposted by Jim Shaw
Now preprinted at arxiv.org/abs/2509.07357

September 10, 2025 at 2:10 AM
Now preprinted at arxiv.org/abs/2509.07357
Reposted by Jim Shaw

How do you long-read sequence metagenomes? I would argue it starts with the right sample storage & DNA extraction, to enable efficient @nanoporetech.com /@pacbio.bsky.social sequencing, which we investigated in our new paper: www.biorxiv.org/content/10.1...
Massive thanks to Klara for driving this
Massive thanks to Klara for driving this

September 9, 2025 at 3:35 PM
How do you long-read sequence metagenomes? I would argue it starts with the right sample storage & DNA extraction, to enable efficient @nanoporetech.com /@pacbio.bsky.social sequencing, which we investigated in our new paper: www.biorxiv.org/content/10.1...
Massive thanks to Klara for driving this
Massive thanks to Klara for driving this
Preprint out for myloasm, our new nanopore / HiFi metagenome assembler!
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
High-resolution metagenome assembly for modern long reads with myloasm https://www.biorxiv.org/content/10.1101/2025.09.05.674543v1
September 7, 2025 at 11:35 PM
Preprint out for myloasm, our new nanopore / HiFi metagenome assembler!
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
Reposted by Jim Shaw

Check out Ryan's new blogpost, especially if you work on and polish small eukaryotic genome assemblies - it's always nice when someone adds new features for your tools
New blog post!
I added a new feature to @gbouras13.bsky.social's Pypolca: homopolymer-only polishing. Potentially useful for cross-sample polishing - early test on Cryptosporidium looks promising.
Check it out here:
rrwick.github.io/2025/09/04/h...
I added a new feature to @gbouras13.bsky.social's Pypolca: homopolymer-only polishing. Potentially useful for cross-sample polishing - early test on Cryptosporidium looks promising.
Check it out here:
rrwick.github.io/2025/09/04/h...
Cross-sample homopolymer polishing with Pypolca
a blog for miscellaneous bioinformatics stuff
rrwick.github.io
September 5, 2025 at 6:20 AM
Check out Ryan's new blogpost, especially if you work on and polish small eukaryotic genome assemblies - it's always nice when someone adds new features for your tools
Reposted by Jim Shaw
Now published in GigaScience with minor improvements: academic.oup.com/gigascience/...
* Download: zenodo.org/records/1490...
* More info: github.com/lh3/panmask
* Download: zenodo.org/records/1490...
* More info: github.com/lh3/panmask
Preprint on "Finding easy regions for short-read variant calling from pangenome data": arxiv.org/abs/2507.03718

September 4, 2025 at 4:44 PM
Now published in GigaScience with minor improvements: academic.oup.com/gigascience/...
* Download: zenodo.org/records/1490...
* More info: github.com/lh3/panmask
* Download: zenodo.org/records/1490...
* More info: github.com/lh3/panmask
Reposted by Jim Shaw

🌎👩🔬 For 15+ years biology has accumulated petabytes (million gigabytes) of🧬DNA sequencing data🧬 from the far reaches of our planet.🦠🍄🌵
Logan now democratizes efficient access to the world’s most comprehensive genetics dataset. Free and open.
doi.org/10.1101/2024...
Logan now democratizes efficient access to the world’s most comprehensive genetics dataset. Free and open.
doi.org/10.1101/2024...

September 3, 2025 at 8:39 AM
🌎👩🔬 For 15+ years biology has accumulated petabytes (million gigabytes) of🧬DNA sequencing data🧬 from the far reaches of our planet.🦠🍄🌵
Logan now democratizes efficient access to the world’s most comprehensive genetics dataset. Free and open.
doi.org/10.1101/2024...
Logan now democratizes efficient access to the world’s most comprehensive genetics dataset. Free and open.
doi.org/10.1101/2024...
Reposted by Jim Shaw

Our study developing a skin metatranscriptomics protocol is now out in @natbiotech.nature.com!
We finally have the ability to study microbial activity on skin and identify key functional genes playing a role in diseases.
Amazing team of Chia Minghao and Amanda Ng 👏
nature.com/articles/s41...
We finally have the ability to study microbial activity on skin and identify key functional genes playing a role in diseases.
Amazing team of Chia Minghao and Amanda Ng 👏
nature.com/articles/s41...
August 30, 2025 at 2:17 AM
Our study developing a skin metatranscriptomics protocol is now out in @natbiotech.nature.com!
We finally have the ability to study microbial activity on skin and identify key functional genes playing a role in diseases.
Amazing team of Chia Minghao and Amanda Ng 👏
nature.com/articles/s41...
We finally have the ability to study microbial activity on skin and identify key functional genes playing a role in diseases.
Amazing team of Chia Minghao and Amanda Ng 👏
nature.com/articles/s41...
Reposted by Jim Shaw

The microbial composition of metagenomes is identified in seconds by profiling against large databases go.nature.com/3BBVqDC
rdcu.be/eCbj4
rdcu.be/eCbj4
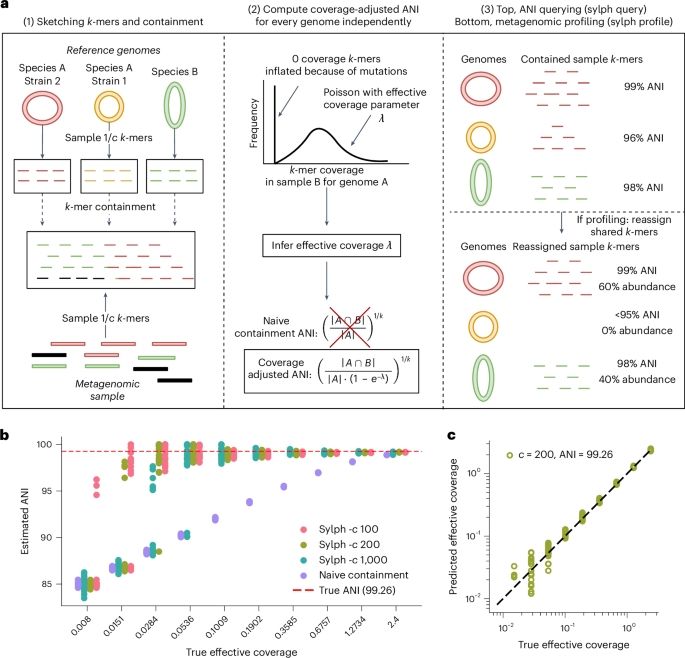
Rapid species-level metagenome profiling and containment estimation with sylph - Nature Biotechnology
The microbial composition of metagenomes is identified in seconds by profiling against large databases.
go.nature.com
August 27, 2025 at 1:14 AM
The microbial composition of metagenomes is identified in seconds by profiling against large databases go.nature.com/3BBVqDC
rdcu.be/eCbj4
rdcu.be/eCbj4
Reposted by Jim Shaw

Thrilled to share our recent review on multidimensional metagenomics analysis, in which we highlight cutting edge technologies and AI applications at 1D to 4D levels. Congrats @hpeng.bsky.social and Angel Ruiz-Moreno.
www.nature.com/articles/s44...
www.nature.com/articles/s44...

Multi-dimensional metagenomics - Nature Reviews Bioengineering
High-throughput sequencing and artificial intelligence-driven structural biology have vastly expanded our understanding of the human metagenome, yet microbial functions remain largely elusive. In this...
www.nature.com
August 26, 2025 at 11:44 AM
Thrilled to share our recent review on multidimensional metagenomics analysis, in which we highlight cutting edge technologies and AI applications at 1D to 4D levels. Congrats @hpeng.bsky.social and Angel Ruiz-Moreno.
www.nature.com/articles/s44...
www.nature.com/articles/s44...
Reposted by Jim Shaw
Our work on direct @nanoporetech.com sequencing of non-canonical bases in now out in @natcomms.nature.com!
Read all about it here: nature.com/articles/s41...
Great collab with Chew and Hirao lab
x.com/NiranjanTW/s...
Read all about it here: nature.com/articles/s41...
Great collab with Chew and Hirao lab
x.com/NiranjanTW/s...

Direct high-throughput deconvolution of non-canonical bases via nanopore sequencing and bootstrapped learning - Nature Communications
Perez, Kimoto, Rajakumar and colleagues present a fast and accurate DNA sequencing method that reads canonical and non-canonical bases using AI and nanopore technology. The approach enables an expande...
nature.com
August 22, 2025 at 9:32 AM
Our work on direct @nanoporetech.com sequencing of non-canonical bases in now out in @natcomms.nature.com!
Read all about it here: nature.com/articles/s41...
Great collab with Chew and Hirao lab
x.com/NiranjanTW/s...
Read all about it here: nature.com/articles/s41...
Great collab with Chew and Hirao lab
x.com/NiranjanTW/s...