



@edg1983.bsky.social
Reposted

Accurate, scalable structural variant genotyping in complex genomes at population scales. #VariantGenotyping #StructuralVariants #PopulationScale #Genomics #Bioinformatics @molbioevol.bsky.social 🧬 🖥️
academic.oup.com/mbe/advance-...
academic.oup.com/mbe/advance-...

August 10, 2025 at 7:32 AM
Accurate, scalable structural variant genotyping in complex genomes at population scales. #VariantGenotyping #StructuralVariants #PopulationScale #Genomics #Bioinformatics @molbioevol.bsky.social 🧬 🖥️
academic.oup.com/mbe/advance-...
academic.oup.com/mbe/advance-...
Reposted
scTail: precise polyadenylation site detection and its alternative usage analysis from reads 1 preserved 3′ scRNA-seq data. #PolyAsite #scRNAseq #GenomeBiology
genomebiology.biomedcentral.com/articles/10....
genomebiology.biomedcentral.com/articles/10....

August 11, 2025 at 9:15 AM
scTail: precise polyadenylation site detection and its alternative usage analysis from reads 1 preserved 3′ scRNA-seq data. #PolyAsite #scRNAseq #GenomeBiology
genomebiology.biomedcentral.com/articles/10....
genomebiology.biomedcentral.com/articles/10....
Reposted

Very excited to share new work from my PhD on a new software package for eQTL mapping: quasar. The quasar software package is a C++ program designed to provide a flexible and efficient eQTL mapping. www.medrxiv.org/content/10.1...

Flexible and efficient count-distribution and mixed-model methods for eQTL mapping with quasar
Identifying genetic variants that affect gene expression, expression quantitative trait loci (eQTLs), is a major focus of modern genomics. Today, various methods exist for eQTL mapping, each using dif...
www.medrxiv.org
July 22, 2025 at 10:15 AM
Very excited to share new work from my PhD on a new software package for eQTL mapping: quasar. The quasar software package is a C++ program designed to provide a flexible and efficient eQTL mapping. www.medrxiv.org/content/10.1...
Reposted
PanVA: a visual analytics tool for pangenomic variant analysis. #Pangenomes #VariantAnalysis #DataVisualization #Genomics #Bioinformatics @biorxiv-bioinfo.bsky.social 🧬 🖥️
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...

June 28, 2025 at 5:05 PM
PanVA: a visual analytics tool for pangenomic variant analysis. #Pangenomes #VariantAnalysis #DataVisualization #Genomics #Bioinformatics @biorxiv-bioinfo.bsky.social 🧬 🖥️
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
Reposted
A Benchmark of Modern Statistical Phasing Methods. #DNAphasing #MethodsBenchmarking #Genomics #Bioinformatics @biorxiv-genomic.bsky.social 🧬 🖥️
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...

June 29, 2025 at 9:15 AM
A Benchmark of Modern Statistical Phasing Methods. #DNAphasing #MethodsBenchmarking #Genomics #Bioinformatics @biorxiv-genomic.bsky.social 🧬 🖥️
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...

Reposted
Preprint on "Improving spliced alignment by modeling splice sites with deep learning". It describes minisplice for modeling splice signals. Minimap2 and miniprot now optionally use the predicted scores to improve spliced alignment.
arxiv.org/abs/2506.12986
arxiv.org/abs/2506.12986
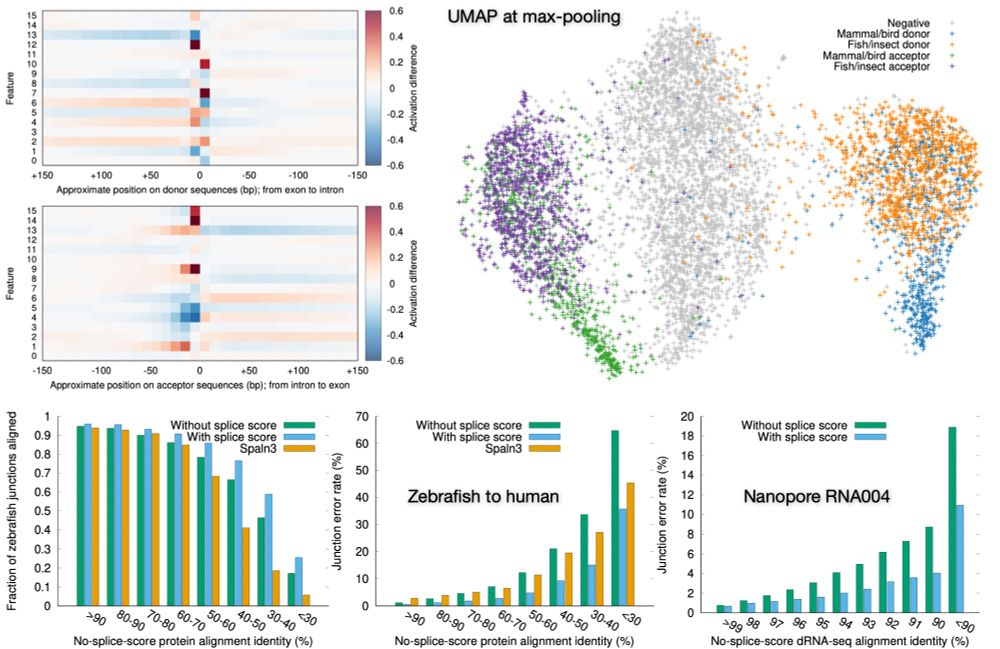
June 17, 2025 at 1:49 AM
Preprint on "Improving spliced alignment by modeling splice sites with deep learning". It describes minisplice for modeling splice signals. Minimap2 and miniprot now optionally use the predicted scores to improve spliced alignment.
arxiv.org/abs/2506.12986
arxiv.org/abs/2506.12986
Reposted

cuteFC: regenotyping structural variants through an accurate and efficient force-calling method genomebiology.biomedcentral.com/articles/10...

June 15, 2025 at 2:15 PM
cuteFC: regenotyping structural variants through an accurate and efficient force-calling method genomebiology.biomedcentral.com/articles/10...
Reposted

📣 Latest from the lab: Performance of deep-learning-based approaches to improve polygenic scores www.nature.com/articles/s41...
Its thought deep learning will substantially improve PGS but the reality is MANY have tried but no/little gain has been seen so far. Here we report our negative results.
Its thought deep learning will substantially improve PGS but the reality is MANY have tried but no/little gain has been seen so far. Here we report our negative results.

June 5, 2025 at 1:37 PM
📣 Latest from the lab: Performance of deep-learning-based approaches to improve polygenic scores www.nature.com/articles/s41...
Its thought deep learning will substantially improve PGS but the reality is MANY have tried but no/little gain has been seen so far. Here we report our negative results.
Its thought deep learning will substantially improve PGS but the reality is MANY have tried but no/little gain has been seen so far. Here we report our negative results.
Reposted

SNP calling, haplotype phasing and allele-specific analysis with long RNA-seq reads www.biorxiv.org/content/10.1... 🧬🖥️🧪
longcallR: github.com/huangnengCSU...
Nextflow workflow: github.com/huangnengCSU...
longcallR: github.com/huangnengCSU...
Nextflow workflow: github.com/huangnengCSU...


May 30, 2025 at 10:29 AM
SNP calling, haplotype phasing and allele-specific analysis with long RNA-seq reads www.biorxiv.org/content/10.1... 🧬🖥️🧪
longcallR: github.com/huangnengCSU...
Nextflow workflow: github.com/huangnengCSU...
longcallR: github.com/huangnengCSU...
Nextflow workflow: github.com/huangnengCSU...
Reposted

Check out our latest blog post to discover how to build custom Docker images for Seqera Studios, with real-world examples including Marimo, Streamlit, CELLxGENE, and Shiny! 🌟
📖 Read the blog post: hubs.la/Q03psL-t0
📖 Read the blog post: hubs.la/Q03psL-t0

May 28, 2025 at 1:29 PM
Check out our latest blog post to discover how to build custom Docker images for Seqera Studios, with real-world examples including Marimo, Streamlit, CELLxGENE, and Shiny! 🌟
📖 Read the blog post: hubs.la/Q03psL-t0
📖 Read the blog post: hubs.la/Q03psL-t0
Reposted

Happy to share our new study on genetic & environmental contributors to age-related decline in ~100K UK Biobank participants!
Here, we used simulation work + longitudinal GWAS and downstream analyses to explore risks involved in cognitive/physical decline
(1/)🧵🧵
shorturl.at/99gqL
Here, we used simulation work + longitudinal GWAS and downstream analyses to explore risks involved in cognitive/physical decline
(1/)🧵🧵
shorturl.at/99gqL

Combining cross-sectional and longitudinal genomic approaches to identify determinants of cognitive and physical decline - Nature Communications
Large-scale genomic studies focusing on the genetic contribution to human aging have mostly relied on cross-sectional data. With the release of longitudinally curated aging phenotypes by the UK Bioban...
shorturl.at
May 19, 2025 at 12:29 PM
Happy to share our new study on genetic & environmental contributors to age-related decline in ~100K UK Biobank participants!
Here, we used simulation work + longitudinal GWAS and downstream analyses to explore risks involved in cognitive/physical decline
(1/)🧵🧵
shorturl.at/99gqL
Here, we used simulation work + longitudinal GWAS and downstream analyses to explore risks involved in cognitive/physical decline
(1/)🧵🧵
shorturl.at/99gqL
Reposted
ConsensuSV-ONT: A modern method for accurate structural variant calling www.nature.com/articles/s41... 🧬🖥️🧪 Nextflow: github.com/SFGLab/Conse...

May 19, 2025 at 3:31 PM
ConsensuSV-ONT: A modern method for accurate structural variant calling www.nature.com/articles/s41... 🧬🖥️🧪 Nextflow: github.com/SFGLab/Conse...
Reposted

🧠 Excited to share my main PhD project! We mapped the regulatory rules governing Glioblastoma plasticity using single-cell multi-omics and deep learning. This work is part of a two-paper series with @bayraktarlab.bsky.social @oliverstegle.bsky.social and @moritzmall.bsky.social, Preprint at end🧵👇
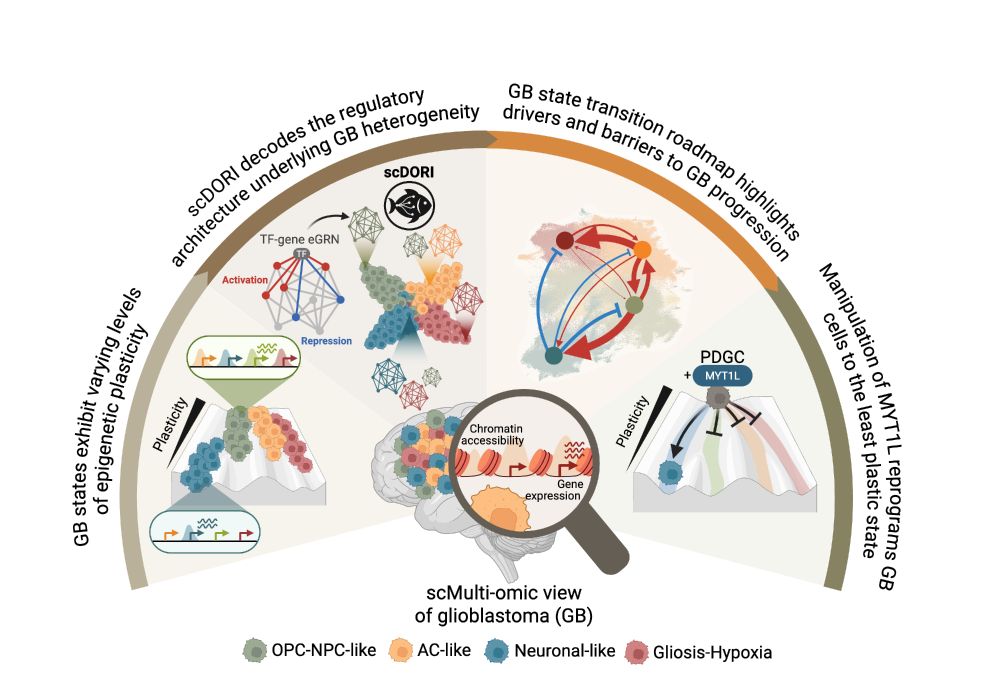
May 16, 2025 at 10:05 AM
🧠 Excited to share my main PhD project! We mapped the regulatory rules governing Glioblastoma plasticity using single-cell multi-omics and deep learning. This work is part of a two-paper series with @bayraktarlab.bsky.social @oliverstegle.bsky.social and @moritzmall.bsky.social, Preprint at end🧵👇
Reposted

📣 Mark your calendars! The 2025 edition of the scverse conference will take place on 17-19 November at Stanford University (US) scverse.org/conference20...
Call for abstracts and registrations coming soon!
Call for abstracts and registrations coming soon!

scverse conference 2025
Follow us on our channels to learn more details in the coming weeks
scverse.org
May 12, 2025 at 10:47 PM
📣 Mark your calendars! The 2025 edition of the scverse conference will take place on 17-19 November at Stanford University (US) scverse.org/conference20...
Call for abstracts and registrations coming soon!
Call for abstracts and registrations coming soon!
Reposted

Our work looking at the effects of using autosomal assumptions for calling variation on X and Y. We used simulated data to compare to a ground truth.
Tldr: Accurate ploidy is important for reducing false positives; appropriate alignment reduces false negatives.
www.biorxiv.org/content/10.1...
Tldr: Accurate ploidy is important for reducing false positives; appropriate alignment reduces false negatives.
www.biorxiv.org/content/10.1...

Best practices for improving alignment and variant calling on human sex chromosomes
Sex chromosome complement is the largest karyotypic variation observed in humans. X and Y chromosomes were once a pair of homologous autosomes. Although chromosome X and Y differentiated from one anot...
www.biorxiv.org
May 7, 2025 at 1:28 PM
Our work looking at the effects of using autosomal assumptions for calling variation on X and Y. We used simulated data to compare to a ground truth.
Tldr: Accurate ploidy is important for reducing false positives; appropriate alignment reduces false negatives.
www.biorxiv.org/content/10.1...
Tldr: Accurate ploidy is important for reducing false positives; appropriate alignment reduces false negatives.
www.biorxiv.org/content/10.1...
Reposted
py_ped_sim: a flexible forward pedigree and genetic simulator for complex family pedigree analysis bmcbioinformatics.biomedcentral.com/articles/10.... 🧬🖥️🧪 github.com/MiguelGuarda...


May 8, 2025 at 6:30 PM
py_ped_sim: a flexible forward pedigree and genetic simulator for complex family pedigree analysis bmcbioinformatics.biomedcentral.com/articles/10.... 🧬🖥️🧪 github.com/MiguelGuarda...
Reposted
Human de novo mutation rates from a four-generation pedigree reference www.nature.com/articles/s41... 🧬🖥️🧪

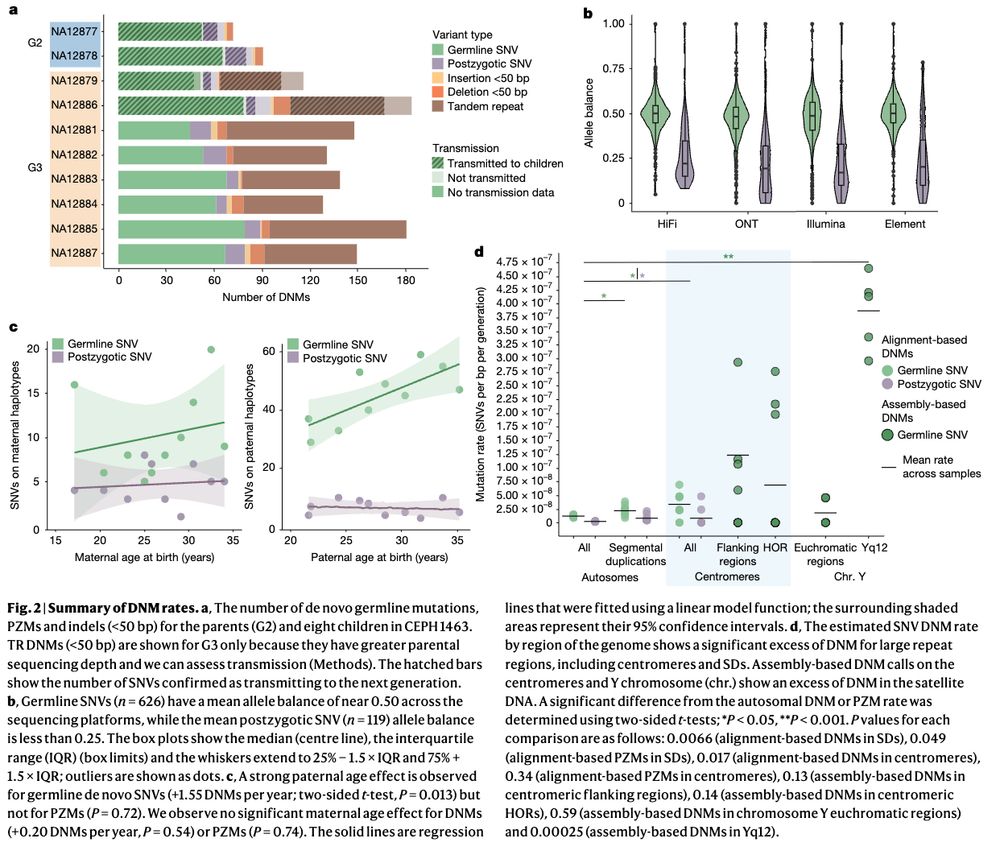


May 2, 2025 at 6:30 PM
Human de novo mutation rates from a four-generation pedigree reference www.nature.com/articles/s41... 🧬🖥️🧪
Reposted

(1/3)🧬 Tissue Genetics: Prevalent genetic effects extend beyond cell boundaries, shaping gut tissue immunology. Work with Aviv Regev, Ramnik Xavier @broadinstitute.org, Kirk Gosik, Heping Xu, Gary Churchill, Kushal K. Dey and other great collaborators.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...

Prevalent cross-cell type QTL trans-regulatory genetic effects impacting innate lymphoid cells in the small intestine
Tissue function in homeostasis and disease arise from coordinated interactions between different cell types and can vary between individuals in a population, in part due to the impact of genetic varia...
www.biorxiv.org
April 20, 2025 at 2:50 PM
(1/3)🧬 Tissue Genetics: Prevalent genetic effects extend beyond cell boundaries, shaping gut tissue immunology. Work with Aviv Regev, Ramnik Xavier @broadinstitute.org, Kirk Gosik, Heping Xu, Gary Churchill, Kushal K. Dey and other great collaborators.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
Reposted

Big updates for the Nextflow @vscode.dev extension! 🚀
🔍 Workflow view – Visualize logical structure of pipelines
⚙️ Process view – See all processes in one place
🌐 Seqera view – Manage connected CEs directly in the IDE
📚 Resources view – Quickly access Copilot, AI tools & training
🔍 Workflow view – Visualize logical structure of pipelines
⚙️ Process view – See all processes in one place
🌐 Seqera view – Manage connected CEs directly in the IDE
📚 Resources view – Quickly access Copilot, AI tools & training
April 17, 2025 at 1:36 PM
Big updates for the Nextflow @vscode.dev extension! 🚀
🔍 Workflow view – Visualize logical structure of pipelines
⚙️ Process view – See all processes in one place
🌐 Seqera view – Manage connected CEs directly in the IDE
📚 Resources view – Quickly access Copilot, AI tools & training
🔍 Workflow view – Visualize logical structure of pipelines
⚙️ Process view – See all processes in one place
🌐 Seqera view – Manage connected CEs directly in the IDE
📚 Resources view – Quickly access Copilot, AI tools & training
Reposted

Some encouraging news for cross-gene generalization of allele effects in S2F models. www.biorxiv.org/content/10.1...

Deep genomic models of allele-specific measurements
Allele-specific quantification of sequencing data, such as gene expression, allows for a causal investigation of how DNA sequence variations influence cis gene regulation. Current methods for analyzin...
www.biorxiv.org
April 16, 2025 at 1:46 AM
Some encouraging news for cross-gene generalization of allele effects in S2F models. www.biorxiv.org/content/10.1...
Reposted

To the top of the "to-read" list. Looks like a heroic amount of work from the Hahn lab (large-scale ChEC-seq compendium!) www.nature.com/articles/s41...
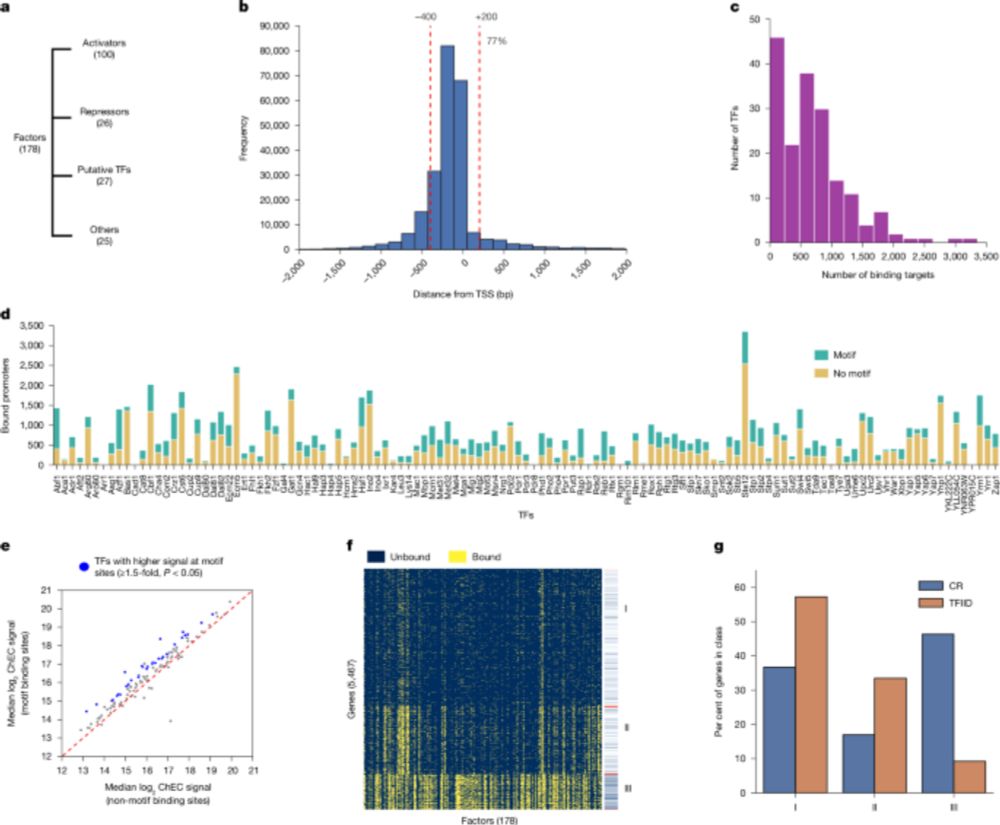
Low overlap of transcription factor DNA binding and regulatory targets - Nature
A near-complete survey of transcription factor activities in Saccharomyces cerevisiae reveals that most transcription factors have both activator and repressor activities and limited overlap between t...
www.nature.com
April 16, 2025 at 4:04 PM
To the top of the "to-read" list. Looks like a heroic amount of work from the Hahn lab (large-scale ChEC-seq compendium!) www.nature.com/articles/s41...
Reposted
New paper out in Genome Biology! 🎉
We lay out best-practice guidelines for releasing variant effect predictors, developed through the Atlas of Variant Effects Alliance @varianteffect.bsky.social
Open, interpretable, and clinically useful VEPs are the goal.
📄 doi.org/10.1186/s130...
We lay out best-practice guidelines for releasing variant effect predictors, developed through the Atlas of Variant Effects Alliance @varianteffect.bsky.social
Open, interpretable, and clinically useful VEPs are the goal.
📄 doi.org/10.1186/s130...

Guidelines for releasing a variant effect predictor - Genome Biology
Computational methods for assessing the likely impacts of mutations, known as variant effect predictors (VEPs), are widely used in the assessment and interpretation of human genetic variation, as well...
doi.org
April 15, 2025 at 12:24 PM
New paper out in Genome Biology! 🎉
We lay out best-practice guidelines for releasing variant effect predictors, developed through the Atlas of Variant Effects Alliance @varianteffect.bsky.social
Open, interpretable, and clinically useful VEPs are the goal.
📄 doi.org/10.1186/s130...
We lay out best-practice guidelines for releasing variant effect predictors, developed through the Atlas of Variant Effects Alliance @varianteffect.bsky.social
Open, interpretable, and clinically useful VEPs are the goal.
📄 doi.org/10.1186/s130...
Reposted
🎙️Just released: Episode 51 of the @nextflow.io podcast!
@ewels.bsky.social and Ben Sherman discuss the upcoming #Nextflow strict syntax - cleaner code, better errors, and a more consistent language framework.
Learn what's changing and how to prepare your pipelines: hubs.la/Q03hpNvx0
@ewels.bsky.social and Ben Sherman discuss the upcoming #Nextflow strict syntax - cleaner code, better errors, and a more consistent language framework.
Learn what's changing and how to prepare your pipelines: hubs.la/Q03hpNvx0

Nextflow strict syntax | The Nextflow Podcast by Seqera
Seqera | The Home for Open Science | From the creators of Nextflow, Wave, and MultiQC
hubs.la
April 15, 2025 at 1:00 PM
🎙️Just released: Episode 51 of the @nextflow.io podcast!
@ewels.bsky.social and Ben Sherman discuss the upcoming #Nextflow strict syntax - cleaner code, better errors, and a more consistent language framework.
Learn what's changing and how to prepare your pipelines: hubs.la/Q03hpNvx0
@ewels.bsky.social and Ben Sherman discuss the upcoming #Nextflow strict syntax - cleaner code, better errors, and a more consistent language framework.
Learn what's changing and how to prepare your pipelines: hubs.la/Q03hpNvx0