




Anthony Nash PhD
@anthonyc1nash.bsky.social
Computational Chemist. Theoretical Biophysicist (physics-based modelling). Protein Dynamics. Unconventional Computing. Metalloproteases.
Pinned

acnash - Overview
Senior Computational Biophysicist and Chemist, Software Engineer, and Medical Statistician. I build novel chemical software to solve protein-disease models. - acnash
github.com
Cleaning up my GitHub page. Most repositories are outdated, and the majority of work has been conducted on private company repositories. Nice picture of me and the dog, though 😅😍
github.com/acnash
github.com/acnash
Good read: www.mdpi.com/2571-9394/6/...
"Data-Centric Benchmarking of Neural Network Architectures for the Univariate Time Series Forecasting Task"
#timeseries #LSTM #realworlddata #neuralnetworks
"Data-Centric Benchmarking of Neural Network Architectures for the Univariate Time Series Forecasting Task"
#timeseries #LSTM #realworlddata #neuralnetworks
www.mdpi.com
September 18, 2025 at 8:23 AM
Good read: www.mdpi.com/2571-9394/6/...
"Data-Centric Benchmarking of Neural Network Architectures for the Univariate Time Series Forecasting Task"
#timeseries #LSTM #realworlddata #neuralnetworks
"Data-Centric Benchmarking of Neural Network Architectures for the Univariate Time Series Forecasting Task"
#timeseries #LSTM #realworlddata #neuralnetworks
Reposted by Anthony Nash PhD
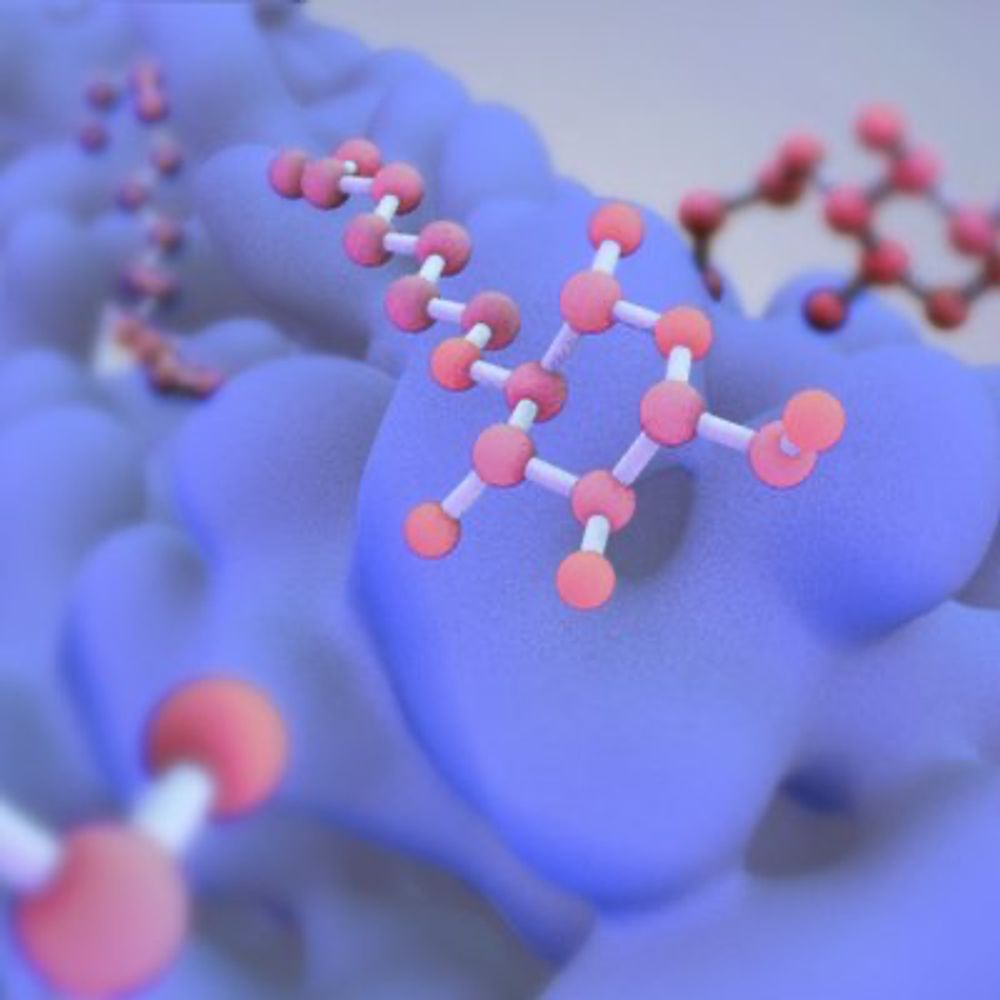
The latest @livecomsjournal.bsky.social tutorial "Molecular Dynamics: From Basics to Application" by Vollmers, Chen et al is out now! doi.org/10.33011/liv...
It includes comprehensive MD tutorials in GROMACS, covering forcefields, thermodynamic ensembles, long-range electrostatics and much more!
It includes comprehensive MD tutorials in GROMACS, covering forcefields, thermodynamic ensembles, long-range electrostatics and much more!

September 9, 2025 at 12:19 PM
The latest @livecomsjournal.bsky.social tutorial "Molecular Dynamics: From Basics to Application" by Vollmers, Chen et al is out now! doi.org/10.33011/liv...
It includes comprehensive MD tutorials in GROMACS, covering forcefields, thermodynamic ensembles, long-range electrostatics and much more!
It includes comprehensive MD tutorials in GROMACS, covering forcefields, thermodynamic ensembles, long-range electrostatics and much more!
Reposted by Anthony Nash PhD

#compchem #compbio Good read: Development of Coarse-Grained Lipid Force Fields Based on a Graph Neural Network pubs.acs.org/doi/10.1021/...

Development of Coarse-Grained Lipid Force Fields Based on a Graph Neural Network
Coarse-grained (CG) lipid models enable efficient simulations of large-scale membrane events. However, achieving both speed and atomic-level accuracy remains challenging. Graph neural networks (GNNs) ...
pubs.acs.org
September 13, 2025 at 7:07 AM
#compchem #compbio Good read: Development of Coarse-Grained Lipid Force Fields Based on a Graph Neural Network pubs.acs.org/doi/10.1021/...
Reposted by Anthony Nash PhD

4️⃣ Featuring the fourth of our showcase projects
Upgrading GROMACS to handle billion-atom systems and enhancing I/O performance and precision, making the first-ever whole-cell simulation possible ➡️ bioexcel.eu/uw67
#MolecularDynamics #GROMACS #ComputationalBiology
Upgrading GROMACS to handle billion-atom systems and enhancing I/O performance and precision, making the first-ever whole-cell simulation possible ➡️ bioexcel.eu/uw67
#MolecularDynamics #GROMACS #ComputationalBiology

September 12, 2025 at 6:22 AM
4️⃣ Featuring the fourth of our showcase projects
Upgrading GROMACS to handle billion-atom systems and enhancing I/O performance and precision, making the first-ever whole-cell simulation possible ➡️ bioexcel.eu/uw67
#MolecularDynamics #GROMACS #ComputationalBiology
Upgrading GROMACS to handle billion-atom systems and enhancing I/O performance and precision, making the first-ever whole-cell simulation possible ➡️ bioexcel.eu/uw67
#MolecularDynamics #GROMACS #ComputationalBiology
Reposted by Anthony Nash PhD

QCxMS2 can now also simulate CID mass spectra.
Just published in #JASMS : doi.org/10.1021/jasms.5c00234
Grateful to my coauthors Stefan Grimme @grimmelab.bsky.social & Marianne Engeser @unibonn.bsky.social - this is the last project of my PhD and completes my work on QCxMS2!
#MassSpec #compchem
Just published in #JASMS : doi.org/10.1021/jasms.5c00234
Grateful to my coauthors Stefan Grimme @grimmelab.bsky.social & Marianne Engeser @unibonn.bsky.social - this is the last project of my PhD and completes my work on QCxMS2!
#MassSpec #compchem
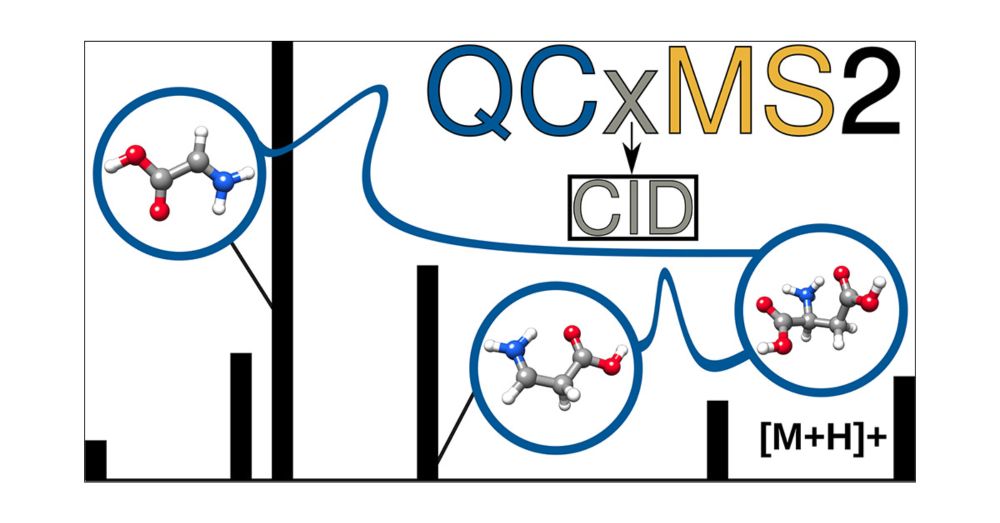
Evaluation of the QCxMS2 Method for the Calculation of Collision-Induced Dissociation Spectra via Automated Reaction Network Exploration
Collision-induced dissociation mass spectrometry (CID-MS) is an important tool in analytical chemistry for the structural elucidation of unknown compounds. The theoretical prediction of the CID spectra plays a critical role in supporting and accelerating this process. To this end, we adapt the recently developed QCxMS2 program originally designed for the calculation of electron ionization (EI) spectra to enable the computation of CID-MS. To account for the fragmentation conditions characteristic of CID within the automated reaction network discovery approach of QCxMS2 we adapted the internal energy distribution to match the experimental conditions. This distribution can be adjusted via a single parameter to approximate various activation settings, thereby eliminating the need for explicit simulations of the collisional process. We evaluate our approach on a test set of 13 organic molecules with diverse functional groups, compiled specifically for this study. All reference spectra were recorded consistently under the same measurement conditions, including both CID and higher-energy collisional dissociation (HCD) modes. Overall, QCxMS2 achieves a good average entropy similarity score (ESS) of 0.687 for the HCD spectra and 0.773 for the CID spectra. The direct comparison to experimental data demonstrates that the QCxMS2 approach, even without explicit modeling of collisions, is generally capable of computing both CID and HCD spectra with reasonable accuracy and robustness. This highlights its potential as a valuable tool for integration into structure elucidation workflows in analytical mass spectrometry.
doi.org
September 8, 2025 at 9:10 AM
QCxMS2 can now also simulate CID mass spectra.
Just published in #JASMS : doi.org/10.1021/jasms.5c00234
Grateful to my coauthors Stefan Grimme @grimmelab.bsky.social & Marianne Engeser @unibonn.bsky.social - this is the last project of my PhD and completes my work on QCxMS2!
#MassSpec #compchem
Just published in #JASMS : doi.org/10.1021/jasms.5c00234
Grateful to my coauthors Stefan Grimme @grimmelab.bsky.social & Marianne Engeser @unibonn.bsky.social - this is the last project of my PhD and completes my work on QCxMS2!
#MassSpec #compchem
Reposted by Anthony Nash PhD

How do proteins really fold? Our latest @pubs.acs.org JPCL paper with @saureli.bsky.social @valeriorizzi.bsky.social @mheritier.bsky.social unveils a new MD strategy to investigate it in atomistic resolution by focusing on water and side-chain interactions. check it out pubs.acs.org/doi/10.1021/...

The Arch from the Stones: Understanding Protein Folding Energy Landscapes via Bioinspired Collective Variables
Protein folding remains a formidable challenge despite significant advances, particularly in sequence-to-structure prediction. Accurately capturing thermodynamics and intermediates via simulations dem...
pubs.acs.org
September 8, 2025 at 1:13 PM
How do proteins really fold? Our latest @pubs.acs.org JPCL paper with @saureli.bsky.social @valeriorizzi.bsky.social @mheritier.bsky.social unveils a new MD strategy to investigate it in atomistic resolution by focusing on water and side-chain interactions. check it out pubs.acs.org/doi/10.1021/...
Reposted by Anthony Nash PhD

Martini 3 Coarse-Grained Models for Carbon Nanomaterials | Journal of Chemical Theory and Computation pubs.acs.org/doi/full/10....

Martini 3 Coarse-Grained Models for Carbon Nanomaterials
The Martini model is a coarse-grained force field allowing simulations of biomolecular systems as well as a range of materials including different types of nanomaterials of technological interest. Recently, a new version of the force field (version 3) has been released that includes new parameters for lipids, proteins, carbohydrates, and a number of small molecules, but not yet carbon nanomaterials. Here, we present new Martini models for three major types of carbon nanomaterials: fullerene, carbon nanotubes, and graphene. The new models were parametrized within the Martini 3 framework, and reproduce semiquantitatively a range of properties for each material. In particular, the model of fullerene yields excellent solid-state properties and good properties in solution, including correct trends in partitioning between different solvents and realistic translocation across lipid membranes. The models of carbon nanotubes reproduce the atomistic behavior of nanotube porins spanning lipid bilayers. The model of graphene reproduces structural and elastic properties, as well as trends in experimental adsorption enthalpies of organic molecules. All new models can be used in large-scale simulations to study the interaction with the wide variety of molecules already available in the Martini 3 force field, including biomolecular and synthetic systems.
pubs.acs.org
September 4, 2025 at 10:07 AM
Martini 3 Coarse-Grained Models for Carbon Nanomaterials | Journal of Chemical Theory and Computation pubs.acs.org/doi/full/10....
There we go... manuscript accepted in Nature.
From now on, I'm painting, playing games, and travelling 😀
From now on, I'm painting, playing games, and travelling 😀
September 2, 2025 at 9:08 PM
There we go... manuscript accepted in Nature.
From now on, I'm painting, playing games, and travelling 😀
From now on, I'm painting, playing games, and travelling 😀
Cleaning up my GitHub page. Most repositories are outdated, and the majority of work has been conducted on private company repositories. Nice picture of me and the dog, though 😅😍
github.com/acnash
github.com/acnash
acnash - Overview
Senior Computational Biophysicist and Chemist, Software Engineer, and Medical Statistician. I build novel chemical software to solve protein-disease models. - acnash
github.com
July 21, 2025 at 6:35 AM
Cleaning up my GitHub page. Most repositories are outdated, and the majority of work has been conducted on private company repositories. Nice picture of me and the dog, though 😅😍
github.com/acnash
github.com/acnash
Reposted by Anthony Nash PhD
#compchem Our recent work "𝐒𝐡𝐨𝐫𝐭𝐜𝐮𝐭 𝐭𝐨 𝐜𝐡𝐞𝐦𝐢𝐜𝐚𝐥𝐥𝐲 𝐚𝐜𝐜𝐮𝐫𝐚𝐭𝐞 𝐪𝐮𝐚𝐧𝐭𝐮𝐦 𝐜𝐨𝐦𝐩𝐮𝐭𝐢𝐧𝐠 𝐯𝐢𝐚 𝐝𝐞𝐧𝐬𝐢𝐭𝐲-𝐛𝐚𝐬𝐞𝐝 𝐛𝐚𝐬𝐢𝐬-𝐬𝐞𝐭 𝐜𝐨𝐫𝐫𝐞𝐜𝐭𝐢𝐨𝐧 " has been selected in the following Nature collection ( #quantumcomputing for Quantum Chemistry section). www.nature.com/collections/...

Methodological developments in electronic structure theory and chemical dynamics
This Collection aims to highlight research that advances our understanding of electronic structure and chemical dynamics, as well as the application of ...
www.nature.com
June 28, 2025 at 11:01 AM
#compchem Our recent work "𝐒𝐡𝐨𝐫𝐭𝐜𝐮𝐭 𝐭𝐨 𝐜𝐡𝐞𝐦𝐢𝐜𝐚𝐥𝐥𝐲 𝐚𝐜𝐜𝐮𝐫𝐚𝐭𝐞 𝐪𝐮𝐚𝐧𝐭𝐮𝐦 𝐜𝐨𝐦𝐩𝐮𝐭𝐢𝐧𝐠 𝐯𝐢𝐚 𝐝𝐞𝐧𝐬𝐢𝐭𝐲-𝐛𝐚𝐬𝐞𝐝 𝐛𝐚𝐬𝐢𝐬-𝐬𝐞𝐭 𝐜𝐨𝐫𝐫𝐞𝐜𝐭𝐢𝐨𝐧 " has been selected in the following Nature collection ( #quantumcomputing for Quantum Chemistry section). www.nature.com/collections/...
I've adjusted the source code of Gaussian accelerated molecular dynamics (GAMD) with OpenMM (github.com/MiaoLab20/ga...) to accept periodic molecules, such as a sequence bonded to itself across the periodic boundary.

June 26, 2025 at 8:48 AM
I've adjusted the source code of Gaussian accelerated molecular dynamics (GAMD) with OpenMM (github.com/MiaoLab20/ga...) to accept periodic molecules, such as a sequence bonded to itself across the periodic boundary.
I'm exploring some software. I check out the dependencies... Perl, MatLab, BLAST, and DSSP.
This is going to break. I just know it.
#sciencesoftware
This is going to break. I just know it.
#sciencesoftware
May 28, 2025 at 11:39 AM
I'm exploring some software. I check out the dependencies... Perl, MatLab, BLAST, and DSSP.
This is going to break. I just know it.
#sciencesoftware
This is going to break. I just know it.
#sciencesoftware
Reposted by Anthony Nash PhD

Our new preprint PharmacoForge: Pharmacophore Generation with Diffusion Models is out now! PharmacoForge quickly generates pharmacophores for a given protein pocket that identify key binding features and find useful compounds in a pharmacophore search. Check it out! 🧪 doi.org/10.26434/che...
May 27, 2025 at 7:11 PM
Our new preprint PharmacoForge: Pharmacophore Generation with Diffusion Models is out now! PharmacoForge quickly generates pharmacophores for a given protein pocket that identify key binding features and find useful compounds in a pharmacophore search. Check it out! 🧪 doi.org/10.26434/che...
I've had to increase the font size used by the favourite IDE. Time stands still for no man.
May 27, 2025 at 4:25 PM
I've had to increase the font size used by the favourite IDE. Time stands still for no man.
Reposted by Anthony Nash PhD

New post: On the failure of rescoring in virtual screening.
Or why automated virtual screening and docking remains hard and why expertise remains essential. medchemash.substack.com/p/on-the-fai...
Or why automated virtual screening and docking remains hard and why expertise remains essential. medchemash.substack.com/p/on-the-fai...

On the failure of rescoring in virtual screening
Why automated virtual screening and docking remains hard and why expertise remains essential
medchemash.substack.com
May 22, 2025 at 7:39 PM
New post: On the failure of rescoring in virtual screening.
Or why automated virtual screening and docking remains hard and why expertise remains essential. medchemash.substack.com/p/on-the-fai...
Or why automated virtual screening and docking remains hard and why expertise remains essential. medchemash.substack.com/p/on-the-fai...
Reposted by Anthony Nash PhD

Now out in @jacs.acspublications.org ! 🎉 : "MACE-OFF: Short-Range Transferable Machine Learning Force Fields for Organic Molecules" by Dávid Kovács, @jhmchem.bsky.social, & team:
pubs.acs.org/doi/10.1021/...
pubs.acs.org/doi/10.1021/...

MACE-OFF: Short-Range Transferable Machine Learning Force Fields for Organic Molecules
Classical empirical force fields have dominated biomolecular simulations for over 50 years. Although widely used in drug discovery, crystal structure prediction, and biomolecular dynamics, they generally lack the accuracy and transferability required for first-principles predictive modeling. In this paper, we introduce MACE-OFF, a series of short-range transferable force fields for organic molecules created using state-of-the-art machine learning technology and first-principles reference data computed with a high level of quantum mechanical theory. MACE-OFF demonstrates the remarkable capabilities of short-range models by accurately predicting a wide variety of gas- and condensed-phase properties of molecular systems. It produces accurate, easy-to-converge dihedral torsion scans of unseen molecules as well as reliable descriptions of molecular crystals and liquids, including quantum nuclear effects. We further demonstrate the capabilities of MACE-OFF by determining free energy surfaces in explicit solvent as well as the folding dynamics of peptides and nanosecond simulations of a fully solvated protein. These developments enable first-principles simulations of molecular systems for the broader chemistry community at high accuracy and relatively low computational cost.
pubs.acs.org
May 19, 2025 at 3:49 PM
Now out in @jacs.acspublications.org ! 🎉 : "MACE-OFF: Short-Range Transferable Machine Learning Force Fields for Organic Molecules" by Dávid Kovács, @jhmchem.bsky.social, & team:
pubs.acs.org/doi/10.1021/...
pubs.acs.org/doi/10.1021/...
Reposted by Anthony Nash PhD

New publication out!
doi.org/10.1093/bioi...
VTX is open source and freely accessible for non commercial use! github.com/VTX-Molecula...
Builds available at vtx.drugdesign.fr
doi.org/10.1093/bioi...
VTX is open source and freely accessible for non commercial use! github.com/VTX-Molecula...
Builds available at vtx.drugdesign.fr
GitHub - VTX-Molecular-Visualization/VTX
Contribute to VTX-Molecular-Visualization/VTX development by creating an account on GitHub.
github.com
May 14, 2025 at 9:59 PM
Reposted by Anthony Nash PhD

In today’s #good_practices #Journal_Club @clarakirkvold.bsky.social discusses the #FAIR #data principles and their implementations in #chemistry www.grynova-ccc.org/journal-club...

May 14, 2025 at 5:45 PM
In today’s #good_practices #Journal_Club @clarakirkvold.bsky.social discusses the #FAIR #data principles and their implementations in #chemistry www.grynova-ccc.org/journal-club...
This is impressive! A huge QM structure of small molecules, ligands, biomolecules, etc., database.
Organisational skills must be at another level.
huggingface.co/facebook/OMo...
And the paper:
arxiv.org/abs/2505.08762
Organisational skills must be at another level.
huggingface.co/facebook/OMo...
And the paper:
arxiv.org/abs/2505.08762

facebook/OMol25 · Hugging Face
We’re on a journey to advance and democratize artificial intelligence through open source and open science.
huggingface.co
May 14, 2025 at 8:59 PM
This is impressive! A huge QM structure of small molecules, ligands, biomolecules, etc., database.
Organisational skills must be at another level.
huggingface.co/facebook/OMo...
And the paper:
arxiv.org/abs/2505.08762
Organisational skills must be at another level.
huggingface.co/facebook/OMo...
And the paper:
arxiv.org/abs/2505.08762
Can you keep up? I sometimes feel like I can't, but remember you're not alone. Just keep reading.
Some tips for performing meaningful and reproducible docking calculations.
journals.plos.org/ploscompbiol...
#docking #moleculardocking #liganddocking #compchem
Some tips for performing meaningful and reproducible docking calculations.
journals.plos.org/ploscompbiol...
#docking #moleculardocking #liganddocking #compchem

Ten quick tips to perform meaningful and reproducible molecular docking calculations
Author summary The ten quick tips presented here are aimed at understanding the drug target thoroughly and performing molecular docking to ensure maximum precision and biological relevance. The emphas...
journals.plos.org
May 12, 2025 at 10:45 AM
Can you keep up? I sometimes feel like I can't, but remember you're not alone. Just keep reading.
Some tips for performing meaningful and reproducible docking calculations.
journals.plos.org/ploscompbiol...
#docking #moleculardocking #liganddocking #compchem
Some tips for performing meaningful and reproducible docking calculations.
journals.plos.org/ploscompbiol...
#docking #moleculardocking #liganddocking #compchem
ML/AI in the sciences is moving at an extraordinary pace. It's easy to feel left behind. Here's an excellent introduction to ML/AI concepts for the experimentalist and theoretician who is frantically reading to keep up.
#AI #ML #machinelearning #science
www.nature.com/articles/s41...
#AI #ML #machinelearning #science
www.nature.com/articles/s41...

A guide to machine learning for biologists - Nature Reviews Molecular Cell Biology
Machine learning is becoming a widely used tool for the analysis of biological data. However, for experimentalists, proper use of machine learning methods can be challenging. This Review provides an o...
www.nature.com
May 12, 2025 at 10:40 AM
ML/AI in the sciences is moving at an extraordinary pace. It's easy to feel left behind. Here's an excellent introduction to ML/AI concepts for the experimentalist and theoretician who is frantically reading to keep up.
#AI #ML #machinelearning #science
www.nature.com/articles/s41...
#AI #ML #machinelearning #science
www.nature.com/articles/s41...
Did you know I'm immortalized in plant form? There is a flower named after me.
Danum Anthony
www.dahliaworld.co.uk/dnamesv.htm#A
Danum Anthony
www.dahliaworld.co.uk/dnamesv.htm#A
Variety name origins
www.dahliaworld.co.uk
May 11, 2025 at 8:39 PM
Did you know I'm immortalized in plant form? There is a flower named after me.
Danum Anthony
www.dahliaworld.co.uk/dnamesv.htm#A
Danum Anthony
www.dahliaworld.co.uk/dnamesv.htm#A
Reposted by Anthony Nash PhD
The CCPBioSim Annual Conference - Frontiers in Biomolecular Simulations will be taking place in Southampton, 14-16 July 2025. Registration is now open! Details and the registration link can be found at www.ccpbiosim.ac.uk/soton2025 #compchem

May 10, 2025 at 12:42 PM
The CCPBioSim Annual Conference - Frontiers in Biomolecular Simulations will be taking place in Southampton, 14-16 July 2025. Registration is now open! Details and the registration link can be found at www.ccpbiosim.ac.uk/soton2025 #compchem
Reposted by Anthony Nash PhD


Reposted by Anthony Nash PhD

Shout-out to Dr. Karla-Sue Marriott and the students at Roger Williams University for using Nanome in their Chemistry of Cannabis course to explore how compounds like THC and Rimonabant interact with CB1 and CB2 cannabinoid receptors in VR! #CannabinoidScience #HigherEdInnovation

May 9, 2025 at 3:05 PM
Shout-out to Dr. Karla-Sue Marriott and the students at Roger Williams University for using Nanome in their Chemistry of Cannabis course to explore how compounds like THC and Rimonabant interact with CB1 and CB2 cannabinoid receptors in VR! #CannabinoidScience #HigherEdInnovation