



Johannes Gorges
@jogorges.bsky.social
PhD student | computational chemist @GrimmeLab @ @UniBonn
(he/him)
(he/him)
QCxMS2 can now also simulate CID mass spectra.
Just published in #JASMS : doi.org/10.1021/jasms.5c00234
Grateful to my coauthors Stefan Grimme @grimmelab.bsky.social & Marianne Engeser @unibonn.bsky.social - this is the last project of my PhD and completes my work on QCxMS2!
#MassSpec #compchem
Just published in #JASMS : doi.org/10.1021/jasms.5c00234
Grateful to my coauthors Stefan Grimme @grimmelab.bsky.social & Marianne Engeser @unibonn.bsky.social - this is the last project of my PhD and completes my work on QCxMS2!
#MassSpec #compchem
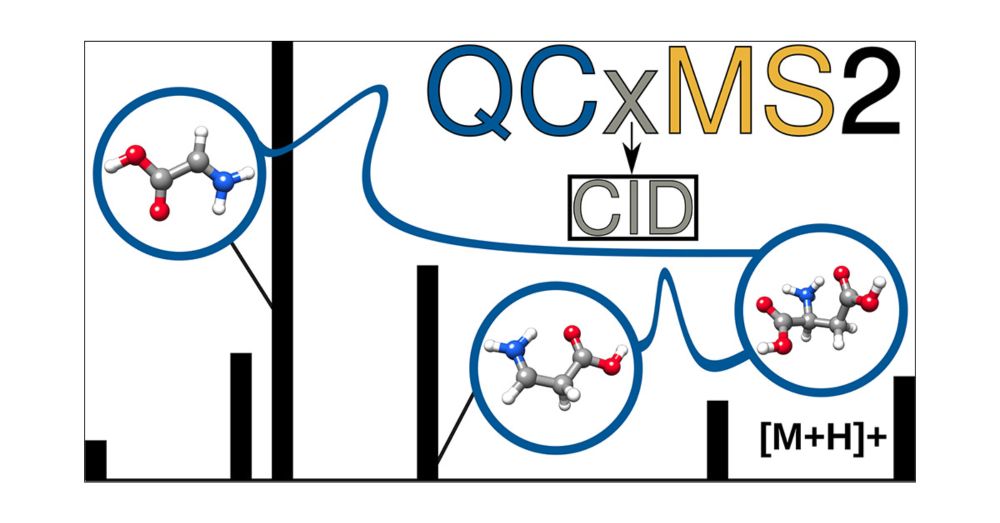
Evaluation of the QCxMS2 Method for the Calculation of Collision-Induced Dissociation Spectra via Automated Reaction Network Exploration
Collision-induced dissociation mass spectrometry (CID-MS) is an important tool in analytical chemistry for the structural elucidation of unknown compounds. The theoretical prediction of the CID spectra plays a critical role in supporting and accelerating this process. To this end, we adapt the recently developed QCxMS2 program originally designed for the calculation of electron ionization (EI) spectra to enable the computation of CID-MS. To account for the fragmentation conditions characteristic of CID within the automated reaction network discovery approach of QCxMS2 we adapted the internal energy distribution to match the experimental conditions. This distribution can be adjusted via a single parameter to approximate various activation settings, thereby eliminating the need for explicit simulations of the collisional process. We evaluate our approach on a test set of 13 organic molecules with diverse functional groups, compiled specifically for this study. All reference spectra were recorded consistently under the same measurement conditions, including both CID and higher-energy collisional dissociation (HCD) modes. Overall, QCxMS2 achieves a good average entropy similarity score (ESS) of 0.687 for the HCD spectra and 0.773 for the CID spectra. The direct comparison to experimental data demonstrates that the QCxMS2 approach, even without explicit modeling of collisions, is generally capable of computing both CID and HCD spectra with reasonable accuracy and robustness. This highlights its potential as a valuable tool for integration into structure elucidation workflows in analytical mass spectrometry.
doi.org
September 8, 2025 at 9:10 AM
QCxMS2 can now also simulate CID mass spectra.
Just published in #JASMS : doi.org/10.1021/jasms.5c00234
Grateful to my coauthors Stefan Grimme @grimmelab.bsky.social & Marianne Engeser @unibonn.bsky.social - this is the last project of my PhD and completes my work on QCxMS2!
#MassSpec #compchem
Just published in #JASMS : doi.org/10.1021/jasms.5c00234
Grateful to my coauthors Stefan Grimme @grimmelab.bsky.social & Marianne Engeser @unibonn.bsky.social - this is the last project of my PhD and completes my work on QCxMS2!
#MassSpec #compchem
Reposted by Johannes Gorges

After almost 3 years of development with @grimmelab.bsky.social, a first preliminary version of our next-generation general extended Tight-Binding (g-xTB) is now on ChemRxiv!
Catch the details at #WATOC: my talk (Thu Session B1) and Stefan’s talk (Thu Session A2).
#compchem
doi.org/10.26434/che...
Catch the details at #WATOC: my talk (Thu Session B1) and Stefan’s talk (Thu Session A2).
#compchem
doi.org/10.26434/che...

g-xTB: A General-Purpose Extended Tight-Binding Electronic Structure Method For the Elements H to Lr (Z=1–103)
We present g-xTB, a next-generation semi-empirical electronic structure method derived from tight-binding (TB) approximations to Kohn–Sham density functional theory (KS-DFT). Designed to bridge the ga...
doi.org
June 24, 2025 at 7:25 AM
After almost 3 years of development with @grimmelab.bsky.social, a first preliminary version of our next-generation general extended Tight-Binding (g-xTB) is now on ChemRxiv!
Catch the details at #WATOC: my talk (Thu Session B1) and Stefan’s talk (Thu Session A2).
#compchem
doi.org/10.26434/che...
Catch the details at #WATOC: my talk (Thu Session B1) and Stefan’s talk (Thu Session A2).
#compchem
doi.org/10.26434/che...
Our paper @grimmelab.bsky.social on QCxMS2 for the calculation of EI-MS was just accepted by @pccp.rsc.org Check out the final article: pubs.rsc.org/en/content/a...
#CompChemSky #MassSpecSky
#CompChemSky #MassSpecSky
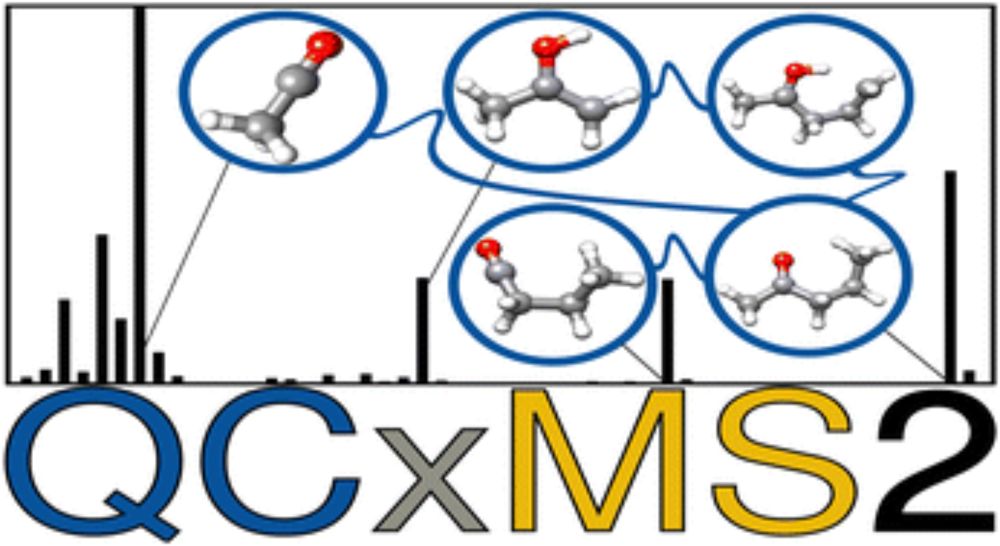
QCxMS2 – a program for the calculation of electron ionization mass spectra via automated reaction network discovery
We present a new fully-automated computational workflow for the calculation of electron ionization mass spectra by automated reaction network discovery, transition state theory and Monte-Carlo simulat...
pubs.rsc.org
March 7, 2025 at 1:08 PM
Our paper @grimmelab.bsky.social on QCxMS2 for the calculation of EI-MS was just accepted by @pccp.rsc.org Check out the final article: pubs.rsc.org/en/content/a...
#CompChemSky #MassSpecSky
#CompChemSky #MassSpecSky
QCxMS2 is here!
Check out our next-generation mass spectra calculation program for EI-MS, based on automated reaction network discovery @grimmelab.bsky.social
-preprint @chemrxiv.bsky.social doi.org/10.26434/che...
-software available at github.com/grimme-lab/Q...
#Massspec #Compchem
Check out our next-generation mass spectra calculation program for EI-MS, based on automated reaction network discovery @grimmelab.bsky.social
-preprint @chemrxiv.bsky.social doi.org/10.26434/che...
-software available at github.com/grimme-lab/Q...
#Massspec #Compchem

GitHub - grimme-lab/QCxMS2: Program package for the quantum mechanical calculation of EI mass spectra using automated reaction network exploration
Program package for the quantum mechanical calculation of EI mass spectra using automated reaction network exploration - grimme-lab/QCxMS2
github.com
February 5, 2025 at 1:45 PM
QCxMS2 is here!
Check out our next-generation mass spectra calculation program for EI-MS, based on automated reaction network discovery @grimmelab.bsky.social
-preprint @chemrxiv.bsky.social doi.org/10.26434/che...
-software available at github.com/grimme-lab/Q...
#Massspec #Compchem
Check out our next-generation mass spectra calculation program for EI-MS, based on automated reaction network discovery @grimmelab.bsky.social
-preprint @chemrxiv.bsky.social doi.org/10.26434/che...
-software available at github.com/grimme-lab/Q...
#Massspec #Compchem
Reposted by Johannes Gorges

Stefan Grimme receives the 2025 Chemistry Europe Award!
Learn more at https://buff.ly/3BXDVhO.
#ChemistryAward #ChemistryEurope
Learn more at https://buff.ly/3BXDVhO.
#ChemistryAward #ChemistryEurope

January 16, 2025 at 7:38 AM
Stefan Grimme receives the 2025 Chemistry Europe Award!
Learn more at https://buff.ly/3BXDVhO.
#ChemistryAward #ChemistryEurope
Learn more at https://buff.ly/3BXDVhO.
#ChemistryAward #ChemistryEurope
See how well low-cost methods describe non-covalent interactions in very large complexes (up to 2000 atoms!) and test your own methods on the LNCI16 benchmark set presented in our article @synlettjournal.bsky.social doi.org/10.1055/s-00... @grimmelab.bsky.social
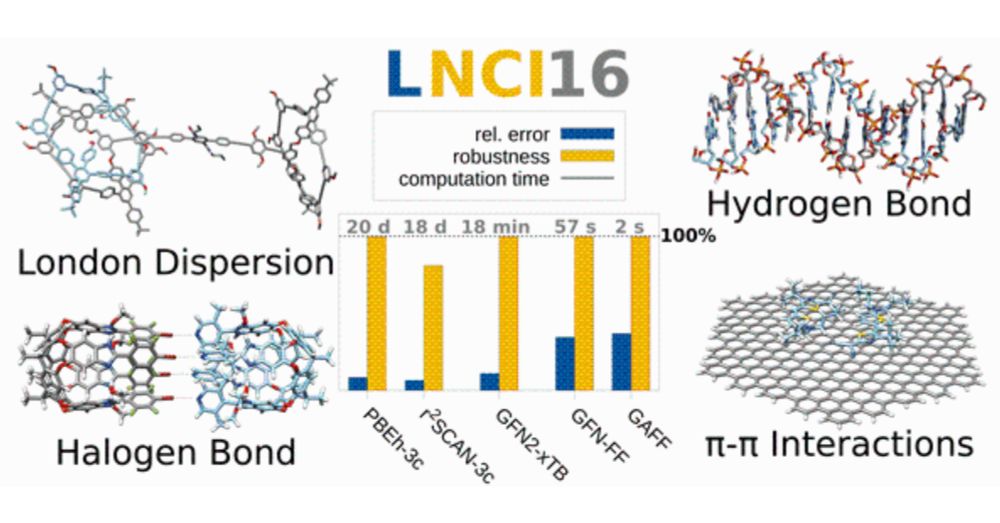
Efficient Computation of the Interaction Energies of Very Large Non-covalently Bound Complexes
Thieme E-Books & E-Journals
doi.org
January 17, 2025 at 4:11 PM
See how well low-cost methods describe non-covalent interactions in very large complexes (up to 2000 atoms!) and test your own methods on the LNCI16 benchmark set presented in our article @synlettjournal.bsky.social doi.org/10.1055/s-00... @grimmelab.bsky.social
If you are interested in computing supramolecular complexes, take a look at this article in @PCCP
and see how well CREST and CENSO work for this challenging task: doi.org/10.1039/D2CP... @grimmelab.bsky.social
#compchem
and see how well CREST and CENSO work for this challenging task: doi.org/10.1039/D2CP... @grimmelab.bsky.social
#compchem
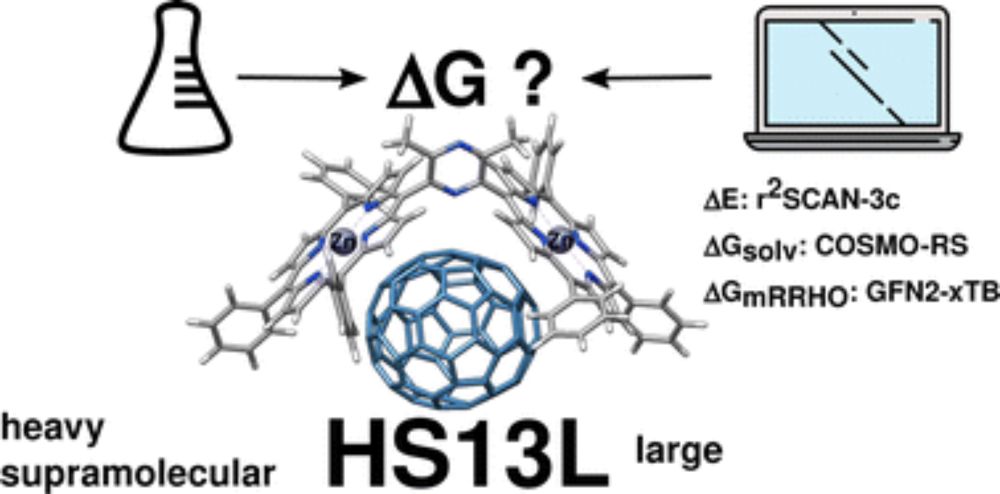
Reliable prediction of association (free) energies of supramolecular complexes with heavy main group elements – the HS13L benchmark set
We introduce a set of 13 supramolecular complexes featuring diverse non-covalent interactions with heavy main group elements (Zn, As, Se, Te, Br, I), high charges (−2 up to +4), and large systems with...
doi.org
January 17, 2025 at 4:08 PM
If you are interested in computing supramolecular complexes, take a look at this article in @PCCP
and see how well CREST and CENSO work for this challenging task: doi.org/10.1039/D2CP... @grimmelab.bsky.social
#compchem
and see how well CREST and CENSO work for this challenging task: doi.org/10.1039/D2CP... @grimmelab.bsky.social
#compchem
Excited to share my first post here on Bluesky! Check out my first paper published in @PCCP, where we @grimmelab.bsky.social investigated the effect of solvation on the conformational #entropy of non-rigid molecules: doi.org/10.1039/D1CP...
#compchem #research
#compchem #research

Towards understanding solvation effects on the conformational entropy of non-rigid molecules
The absolute molecular entropy is a fundamental quantity for the accurate description of thermodynamic properties. For non-rigid molecules, a substantial part of the entropy can be attributed to a con...
doi.org
January 17, 2025 at 4:06 PM
Excited to share my first post here on Bluesky! Check out my first paper published in @PCCP, where we @grimmelab.bsky.social investigated the effect of solvation on the conformational #entropy of non-rigid molecules: doi.org/10.1039/D1CP...
#compchem #research
#compchem #research