




SecureBio, Inc.
@securebio.org
We modeled how such a system could integrate metagenomic sequencing to detect both known and novel pathogens: naobservatory.org/blog/biothreat_radar

November 12, 2025 at 6:30 PM
We modeled how such a system could integrate metagenomic sequencing to detect both known and novel pathogens: naobservatory.org/blog/biothreat_radar
Our paper on the sensitivity of wastewater metagenomic sequencing for early detection of viruses has now been published in The Lancet Microbe: www.thelancet.com/journals/lan...

November 12, 2025 at 6:30 PM
Our paper on the sensitivity of wastewater metagenomic sequencing for early detection of viruses has now been published in The Lancet Microbe: www.thelancet.com/journals/lan...
By now, our detection system frequently flags suspicious reads. But it doesn't recover the surrounding genome.
Earlier this year, we showed how our outward assembly pipeline can recover these genomes, testing the algorithm on a flagged SARS-CoV-2/plasmid construct where it did extremely well.
Earlier this year, we showed how our outward assembly pipeline can recover these genomes, testing the algorithm on a flagged SARS-CoV-2/plasmid construct where it did extremely well.

November 12, 2025 at 6:30 PM
By now, our detection system frequently flags suspicious reads. But it doesn't recover the surrounding genome.
Earlier this year, we showed how our outward assembly pipeline can recover these genomes, testing the algorithm on a flagged SARS-CoV-2/plasmid construct where it did extremely well.
Earlier this year, we showed how our outward assembly pipeline can recover these genomes, testing the algorithm on a flagged SARS-CoV-2/plasmid construct where it did extremely well.
We’ve enhanced our data analysis systems, scaling metadata management, improving automated detection pipelines, and beginning integration of frontier LLMs for automated viral flag analysis.

November 12, 2025 at 6:30 PM
We’ve enhanced our data analysis systems, scaling metadata management, improving automated detection pipelines, and beginning integration of frontier LLMs for automated viral flag analysis.
Zephyr, our Boston-based swab sampling program, has also expanded.
With four dedicated field samplers working most weekdays, we collect 300-800 nasal swabs weekly.
Viral reads are available via our dashboard: data.securebio.org/sampling-met...
With four dedicated field samplers working most weekdays, we collect 300-800 nasal swabs weekly.
Viral reads are available via our dashboard: data.securebio.org/sampling-met...

November 12, 2025 at 6:30 PM
Zephyr, our Boston-based swab sampling program, has also expanded.
With four dedicated field samplers working most weekdays, we collect 300-800 nasal swabs weekly.
Viral reads are available via our dashboard: data.securebio.org/sampling-met...
With four dedicated field samplers working most weekdays, we collect 300-800 nasal swabs weekly.
Viral reads are available via our dashboard: data.securebio.org/sampling-met...
Nucleic Acid Observatory updates:
- Major wastewater surveillance scaling: expanded to 31 sampling sites across 19 cities.
- Zephyr swab program scaling, now 400-800 swabs per week.
- New team members that lead response, wet-lab, and partnerships work.
- Major wastewater surveillance scaling: expanded to 31 sampling sites across 19 cities.
- Zephyr swab program scaling, now 400-800 swabs per week.
- New team members that lead response, wet-lab, and partnerships work.

November 12, 2025 at 6:30 PM
Nucleic Acid Observatory updates:
- Major wastewater surveillance scaling: expanded to 31 sampling sites across 19 cities.
- Zephyr swab program scaling, now 400-800 swabs per week.
- New team members that lead response, wet-lab, and partnerships work.
- Major wastewater surveillance scaling: expanded to 31 sampling sites across 19 cities.
- Zephyr swab program scaling, now 400-800 swabs per week.
- New team members that lead response, wet-lab, and partnerships work.
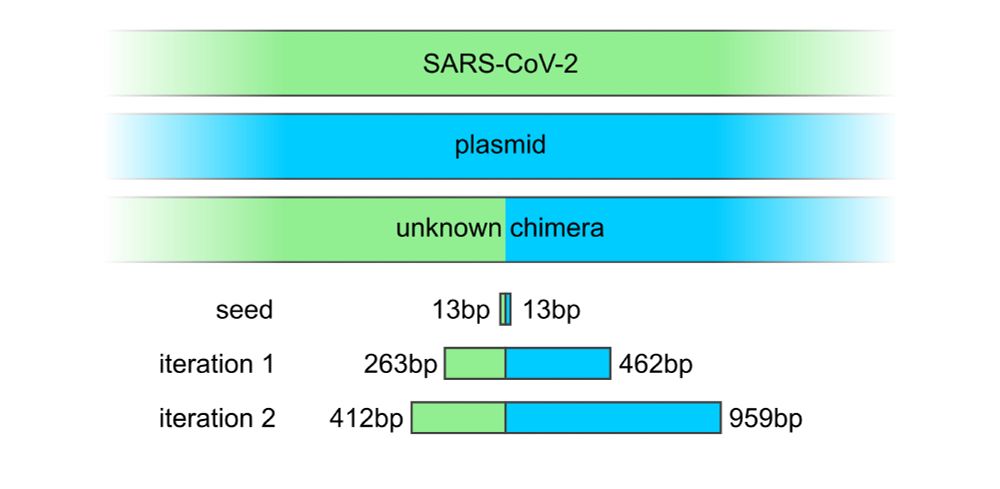
SecureBio’s NAO detection system flags suspicious reads, but doesn't recover the surrounding genome.
We can recover these genomes with our outward assembly pipeline.
We recently tested outward assembly on a flagged SARS-CoV-2/plasmid construct – it did extremely well!
We can recover these genomes with our outward assembly pipeline.
We recently tested outward assembly on a flagged SARS-CoV-2/plasmid construct – it did extremely well!
July 25, 2025 at 4:32 PM
SecureBio’s NAO detection system flags suspicious reads, but doesn't recover the surrounding genome.
We can recover these genomes with our outward assembly pipeline.
We recently tested outward assembly on a flagged SARS-CoV-2/plasmid construct – it did extremely well!
We can recover these genomes with our outward assembly pipeline.
We recently tested outward assembly on a flagged SARS-CoV-2/plasmid construct – it did extremely well!
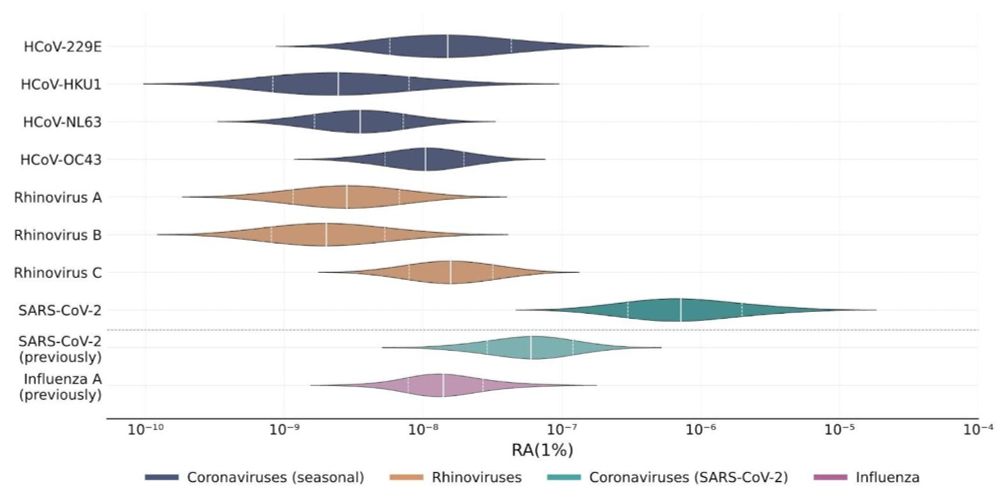
SARS-CoV-2 is highly detectable, but cold viruses are harder to detect.
We will refine these estimates as we generate more sequencing data.
naobservatory.org/blog/swab-ba...
We will refine these estimates as we generate more sequencing data.
naobservatory.org/blog/swab-ba...
July 10, 2025 at 9:31 PM
SARS-CoV-2 is highly detectable, but cold viruses are harder to detect.
We will refine these estimates as we generate more sequencing data.
naobservatory.org/blog/swab-ba...
We will refine these estimates as we generate more sequencing data.
naobservatory.org/blog/swab-ba...
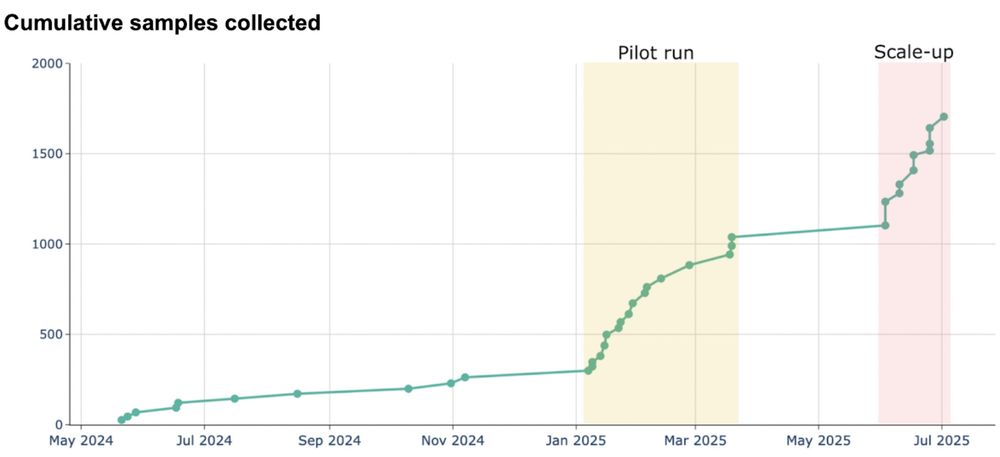
After our sampling sprint earlier this year, we’ve been scaling our Boston-based swab sampling program, hiring several field samplers.
This has allowed us to collect 100-200 swabs per day. Over the coming quarter we will scale to additional weekdays and further optimize sampling.
This has allowed us to collect 100-200 swabs per day. Over the coming quarter we will scale to additional weekdays and further optimize sampling.
July 10, 2025 at 9:31 PM
After our sampling sprint earlier this year, we’ve been scaling our Boston-based swab sampling program, hiring several field samplers.
This has allowed us to collect 100-200 swabs per day. Over the coming quarter we will scale to additional weekdays and further optimize sampling.
This has allowed us to collect 100-200 swabs per day. Over the coming quarter we will scale to additional weekdays and further optimize sampling.
Our partners @solidevidence and Dave O'Connor have created new dashboards that present which pathogens we see in wastewater.
Find the dashboard here: dholab.github.io/public_viz/
Find the dashboard here: dholab.github.io/public_viz/
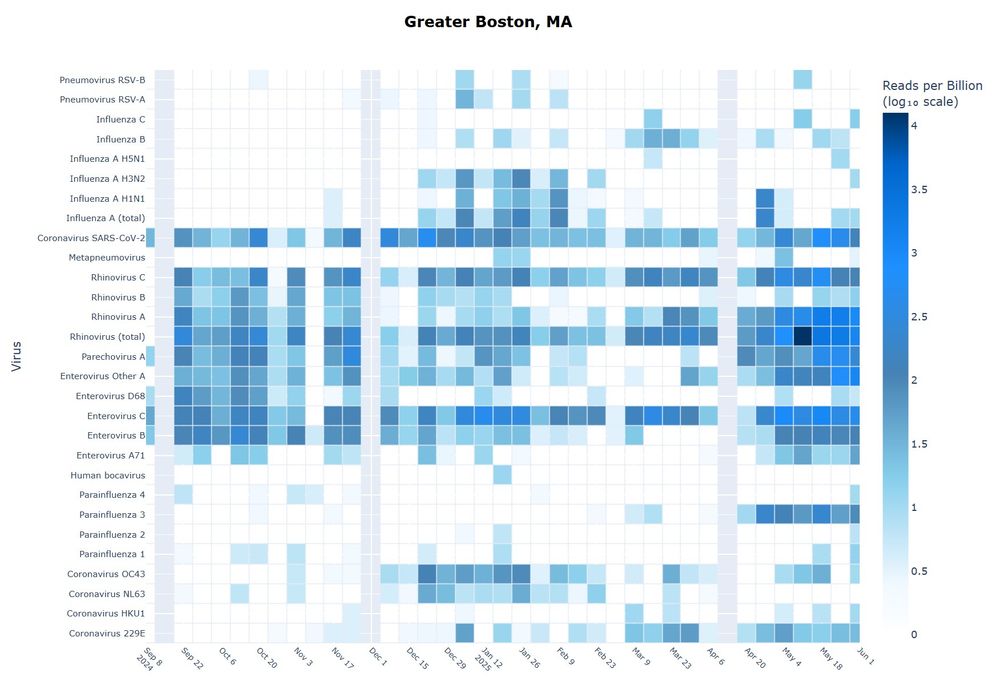
July 10, 2025 at 9:31 PM
Our partners @solidevidence and Dave O'Connor have created new dashboards that present which pathogens we see in wastewater.
Find the dashboard here: dholab.github.io/public_viz/
Find the dashboard here: dholab.github.io/public_viz/
We’ve generated another 487B read pairs, through both our own sequencing, and through Marc Johnson’s lab (@solidevidence).
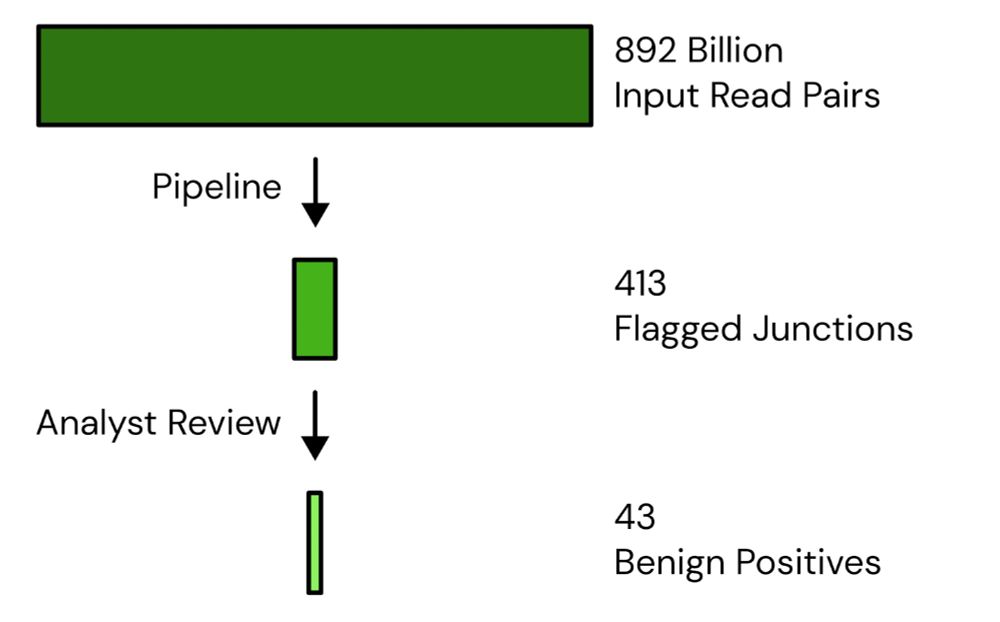
With the new data, our genetic engineering detection system has now analyzed 892B read pairs, flagging 413 chimeras, with 43 “benign positives”.
With the new data, our genetic engineering detection system has now analyzed 892B read pairs, flagging 413 chimeras, with 43 “benign positives”.
July 10, 2025 at 9:31 PM
We’ve generated another 487B read pairs, through both our own sequencing, and through Marc Johnson’s lab (@solidevidence).
With the new data, our genetic engineering detection system has now analyzed 892B read pairs, flagging 413 chimeras, with 43 “benign positives”.
With the new data, our genetic engineering detection system has now analyzed 892B read pairs, flagging 413 chimeras, with 43 “benign positives”.
Nucleic Acid Observatory updates:
- We’ve further increased our sequencing capacity, producing 487B read pairs.
- Our partners created dashboards that summarize which pathogens we routinely see in wastewater.
- We’ve been scaling up our Boston-based swab sampling program.
- We’ve further increased our sequencing capacity, producing 487B read pairs.
- Our partners created dashboards that summarize which pathogens we routinely see in wastewater.
- We’ve been scaling up our Boston-based swab sampling program.

July 10, 2025 at 9:31 PM
Nucleic Acid Observatory updates:
- We’ve further increased our sequencing capacity, producing 487B read pairs.
- Our partners created dashboards that summarize which pathogens we routinely see in wastewater.
- We’ve been scaling up our Boston-based swab sampling program.
- We’ve further increased our sequencing capacity, producing 487B read pairs.
- Our partners created dashboards that summarize which pathogens we routinely see in wastewater.
- We’ve been scaling up our Boston-based swab sampling program.
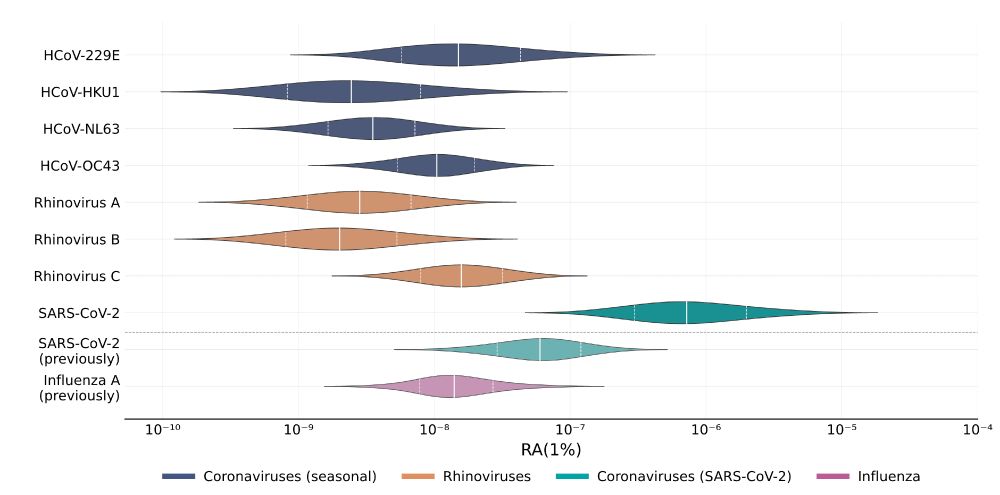
Linking this prevalence data with wastewater data, we estimated how easily different pathogens are detected in sequenced sewage.
SARS-CoV-2 was again confirmed to be readily detectable, but cold viruses showed relatively low detectability.
SARS-CoV-2 was again confirmed to be readily detectable, but cold viruses showed relatively low detectability.
June 26, 2025 at 1:50 PM
Linking this prevalence data with wastewater data, we estimated how easily different pathogens are detected in sequenced sewage.
SARS-CoV-2 was again confirmed to be readily detectable, but cold viruses showed relatively low detectability.
SARS-CoV-2 was again confirmed to be readily detectable, but cold viruses showed relatively low detectability.
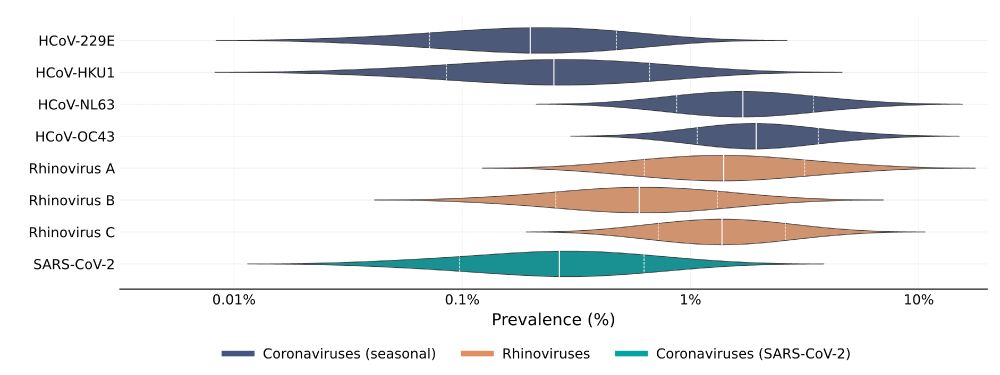
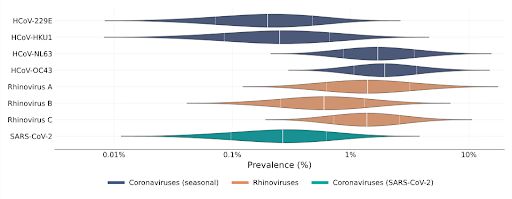
Using this we were able to estimate prevalence.
We found that cold viruses are highly prevalent in winter: Many have a prevalence of 1% or higher!
We found that cold viruses are highly prevalent in winter: Many have a prevalence of 1% or higher!
June 26, 2025 at 1:50 PM
Using this we were able to estimate prevalence.
We found that cold viruses are highly prevalent in winter: Many have a prevalence of 1% or higher!
We found that cold viruses are highly prevalent in winter: Many have a prevalence of 1% or higher!
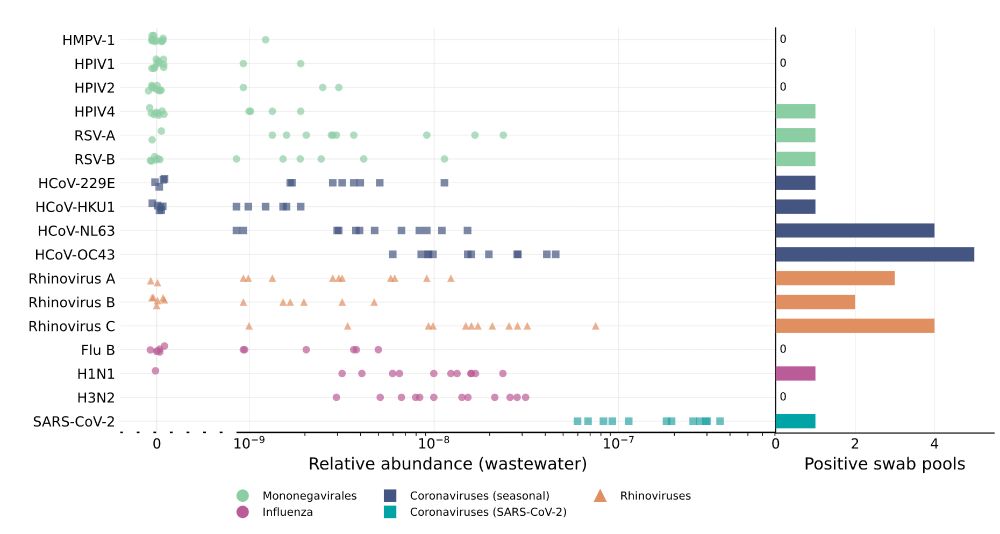
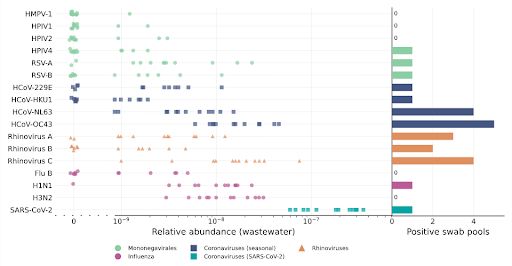
We created these estimates by analyzing nasal swabs, a recently added NAO sample stream.
Collecting, pooling, and sequencing nasal swabs, we got a lot of information about the presence of cold viruses in the population, which we then linked with paired wastewater data.
Collecting, pooling, and sequencing nasal swabs, we got a lot of information about the presence of cold viruses in the population, which we then linked with paired wastewater data.
June 26, 2025 at 1:50 PM
We created these estimates by analyzing nasal swabs, a recently added NAO sample stream.
Collecting, pooling, and sequencing nasal swabs, we got a lot of information about the presence of cold viruses in the population, which we then linked with paired wastewater data.
Collecting, pooling, and sequencing nasal swabs, we got a lot of information about the presence of cold viruses in the population, which we then linked with paired wastewater data.
At the NAO, we’ve created new estimates on how well wastewater sequencing detects different pathogens.
SARS-CoV-2 is again highly detectable, but common cold viruses are harder to detect.
We will use this research to compare wastewater sequencing to other detection strategies.
SARS-CoV-2 is again highly detectable, but common cold viruses are harder to detect.
We will use this research to compare wastewater sequencing to other detection strategies.

June 26, 2025 at 1:50 PM
At the NAO, we’ve created new estimates on how well wastewater sequencing detects different pathogens.
SARS-CoV-2 is again highly detectable, but common cold viruses are harder to detect.
We will use this research to compare wastewater sequencing to other detection strategies.
SARS-CoV-2 is again highly detectable, but common cold viruses are harder to detect.
We will use this research to compare wastewater sequencing to other detection strategies.
Using this we were able to estimate prevalence. We found that cold viruses are highly prevalent in winter: Many have a prevalence of 1% or higher!
June 19, 2025 at 7:18 PM
Using this we were able to estimate prevalence. We found that cold viruses are highly prevalent in winter: Many have a prevalence of 1% or higher!
We created these estimates by analyzing nasal swabs, a recently added NAO sample stream. Collecting, pooling, and sequencing nasal swabs, we got a lot of information about the presence of cold viruses in the population, which we then linked with paired wastewater data.
June 19, 2025 at 7:18 PM
We created these estimates by analyzing nasal swabs, a recently added NAO sample stream. Collecting, pooling, and sequencing nasal swabs, we got a lot of information about the presence of cold viruses in the population, which we then linked with paired wastewater data.
SecureBio, partnering with Ginkgo Biosecurity will host an evening event on how to accelerate biosecurity on Thursday, July 31st, from 6:00-8:30 pm, in Boston (sign-up link in the next post).

June 18, 2025 at 7:06 PM
SecureBio, partnering with Ginkgo Biosecurity will host an evening event on how to accelerate biosecurity on Thursday, July 31st, from 6:00-8:30 pm, in Boston (sign-up link in the next post).
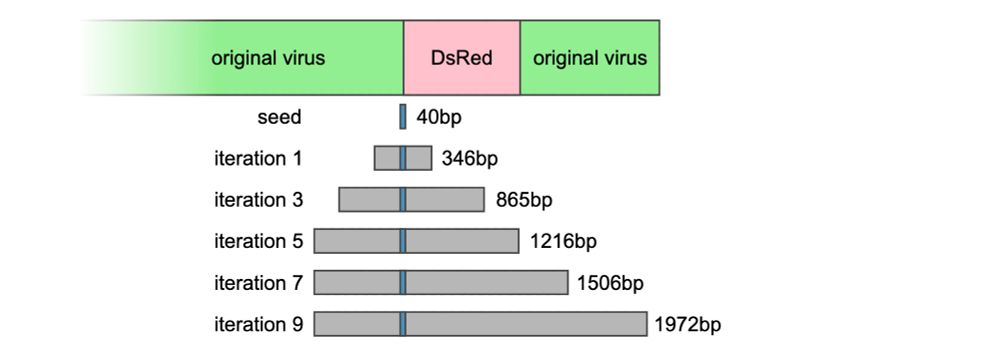
Standard metagenomic assembly at our scale would be prohibitively expensive.
Instead, our approach searches billions of reads to find only those relevant to the seed, then assembles just those reads. 3/4
Instead, our approach searches billions of reads to find only those relevant to the seed, then assembles just those reads. 3/4
April 25, 2025 at 5:28 PM
Standard metagenomic assembly at our scale would be prohibitively expensive.
Instead, our approach searches billions of reads to find only those relevant to the seed, then assembles just those reads. 3/4
Instead, our approach searches billions of reads to find only those relevant to the seed, then assembles just those reads. 3/4
We've now developed outward assembly—a new open-source pipeline that efficiently builds longer sequences by extending outward from a suspicious "seed" sequence, like a read section flagged by our chimera detection pipeline. 2/4

April 25, 2025 at 5:28 PM
We've now developed outward assembly—a new open-source pipeline that efficiently builds longer sequences by extending outward from a suspicious "seed" sequence, like a read section flagged by our chimera detection pipeline. 2/4
Here is a sample of the results. Each dot is a set of questions. The y-axis is a model's score on that set minus the score of the virologist for which the set was tailored. Many models outperform most experts, with o3 exceeding 94% of them. 10/13

April 23, 2025 at 9:51 PM
Here is a sample of the results. Each dot is a set of questions. The y-axis is a model's score on that set minus the score of the virologist for which the set was tailored. Many models outperform most experts, with o3 exceeding 94% of them. 10/13
Can AIs provide expert-level troubleshooting assistance for work with viruses?
We built a new benchmark to answer that question.
To our surprise, we found that leading models outperform the vast majority of practicing virologists we sampled. 🧵 1/13
We built a new benchmark to answer that question.
To our surprise, we found that leading models outperform the vast majority of practicing virologists we sampled. 🧵 1/13

April 23, 2025 at 9:51 PM
Can AIs provide expert-level troubleshooting assistance for work with viruses?
We built a new benchmark to answer that question.
To our surprise, we found that leading models outperform the vast majority of practicing virologists we sampled. 🧵 1/13
We built a new benchmark to answer that question.
To our surprise, we found that leading models outperform the vast majority of practicing virologists we sampled. 🧵 1/13