



@isidrolauscher.bsky.social
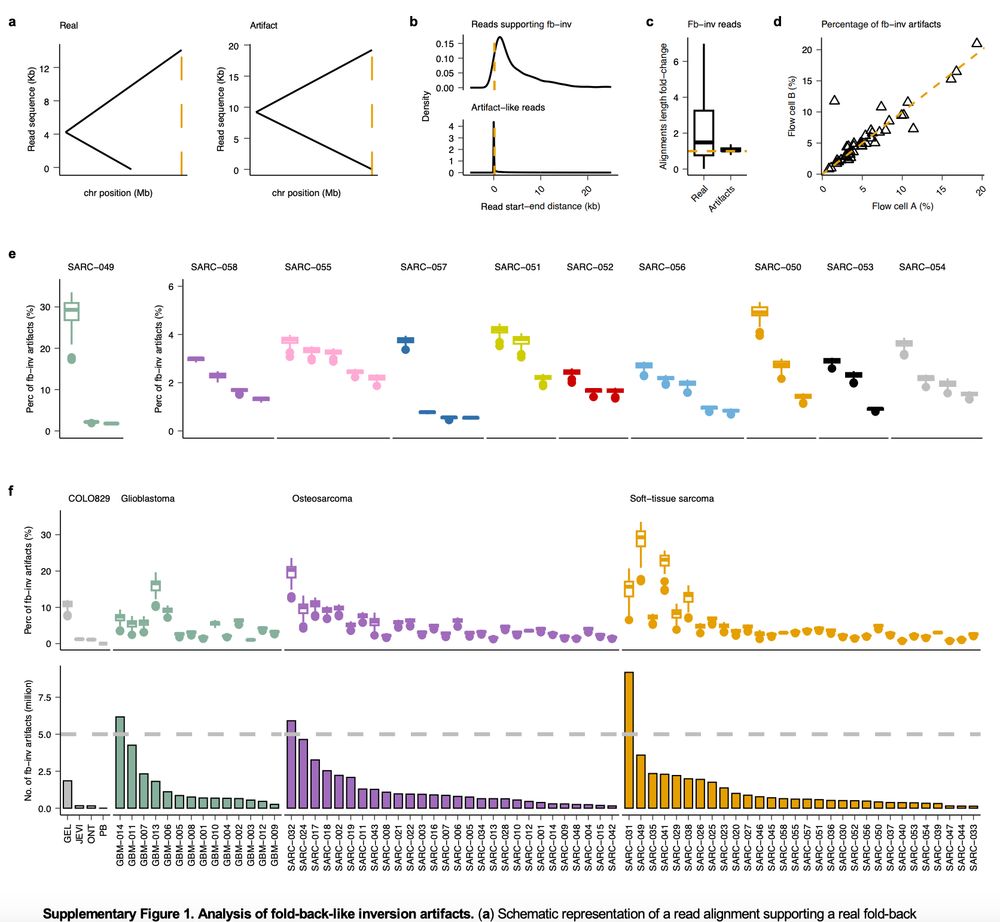
Very glad to see this preprint by @lh3lh3.bsky.social and Meyerson labs www.biorxiv.org/content/10.1... confirming our finding of artifactual fold-back inv in long reads (Fig S1 in our @natmethods.nature.com paper presenting SAVANA, which filters such artifacts to improve SV calling 👇
July 20, 2025 at 11:35 PM
Very glad to see this preprint by @lh3lh3.bsky.social and Meyerson labs www.biorxiv.org/content/10.1... confirming our finding of artifactual fold-back inv in long reads (Fig S1 in our @natmethods.nature.com paper presenting SAVANA, which filters such artifacts to improve SV calling 👇
In sum, we establish best practices for benchmarking SV detection methods for somatic (eg cancer) genome analysis, and show that SAVANA enables the application of long-read sequencing to detect SVs and SCNAs reliably in clinical samples.
May 28, 2025 at 5:36 PM
In sum, we establish best practices for benchmarking SV detection methods for somatic (eg cancer) genome analysis, and show that SAVANA enables the application of long-read sequencing to detect SVs and SCNAs reliably in clinical samples.
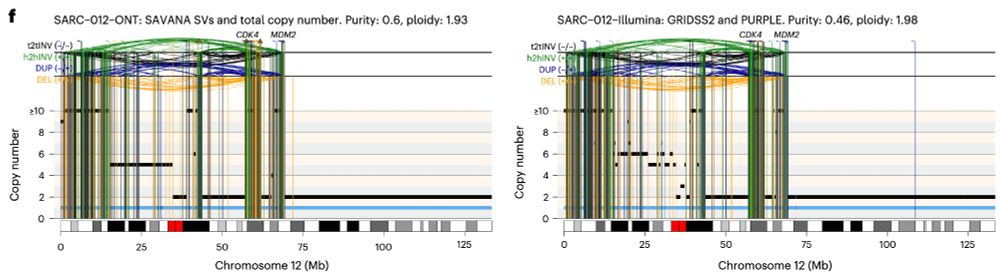
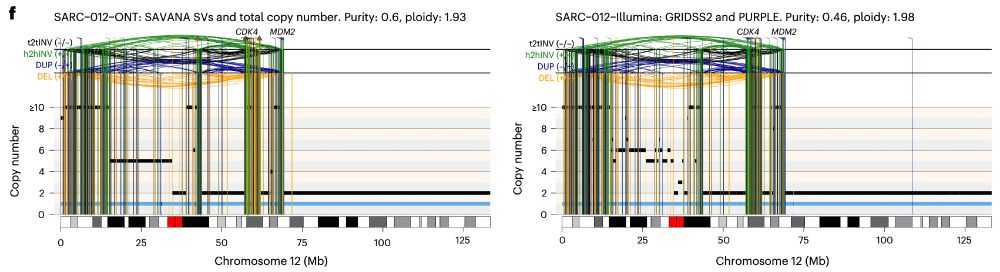
In practice, this means that we can now study, reliably, complex genomic rearrangements (e.g. #chromothripsis) and clinically relevant events causing tumour suppressor gene loss using long reads (left) with comparable accuracy to Illumina (right):
May 28, 2025 at 5:36 PM
In practice, this means that we can now study, reliably, complex genomic rearrangements (e.g. #chromothripsis) and clinically relevant events causing tumour suppressor gene loss using long reads (left) with comparable accuracy to Illumina (right):
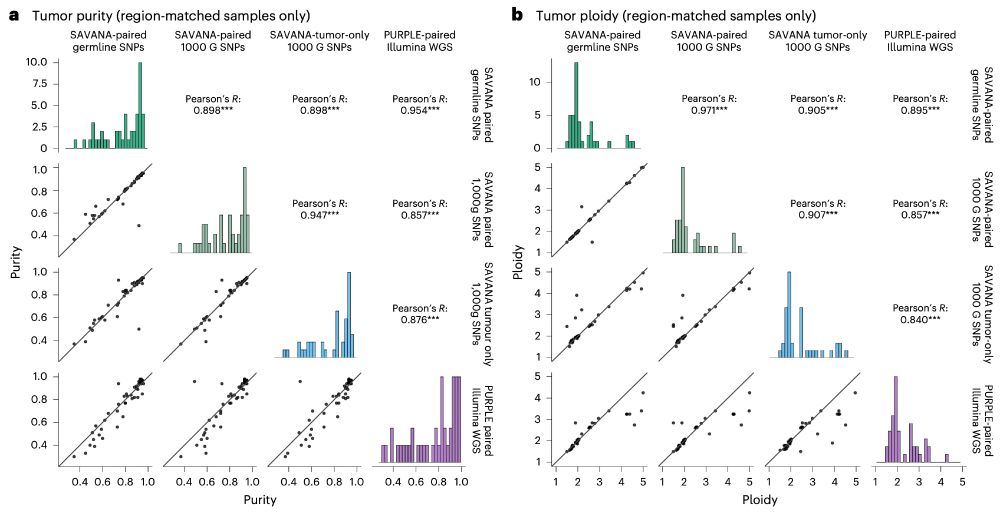
Moreover, using #SAVANA, we can estimate tumour purity and ploidy with comparable accuracy to illumina data (using the fantastic pipeline developed by the Hartwig Medical Foundation @ecuppen.bsky.social @danielisskeptical.bsky.social for clinical reports) even WITHOUT a germline control!
May 28, 2025 at 5:36 PM
Moreover, using #SAVANA, we can estimate tumour purity and ploidy with comparable accuracy to illumina data (using the fantastic pipeline developed by the Hartwig Medical Foundation @ecuppen.bsky.social @danielisskeptical.bsky.social for clinical reports) even WITHOUT a germline control!
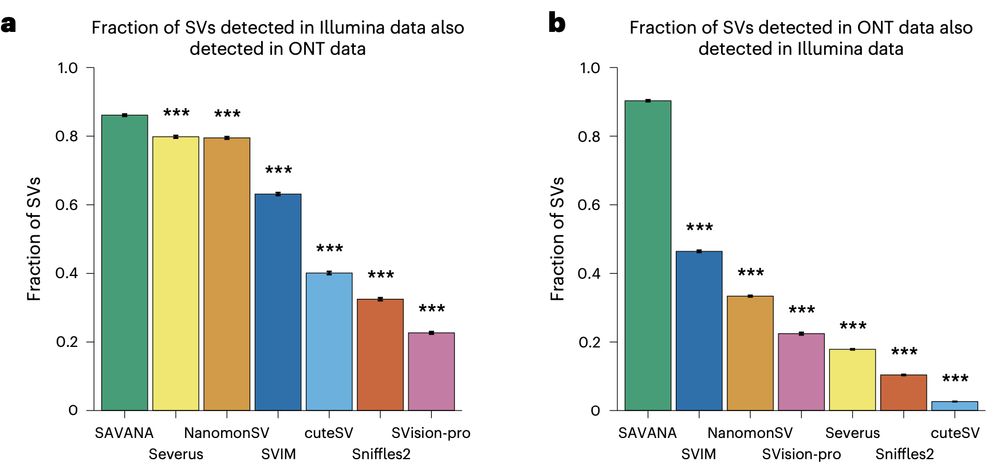
Using SAVANA, we recover most of the SVs detected in short-read data (note the higher than two-fold diff in coverage between long and short reads here!!), and most of the SVs detected using long reads are detected in illumina data (note that we are not using ultra-long reads)
May 28, 2025 at 5:36 PM
Using SAVANA, we recover most of the SVs detected in short-read data (note the higher than two-fold diff in coverage between long and short reads here!!), and most of the SVs detected using long reads are detected in illumina data (note that we are not using ultra-long reads)
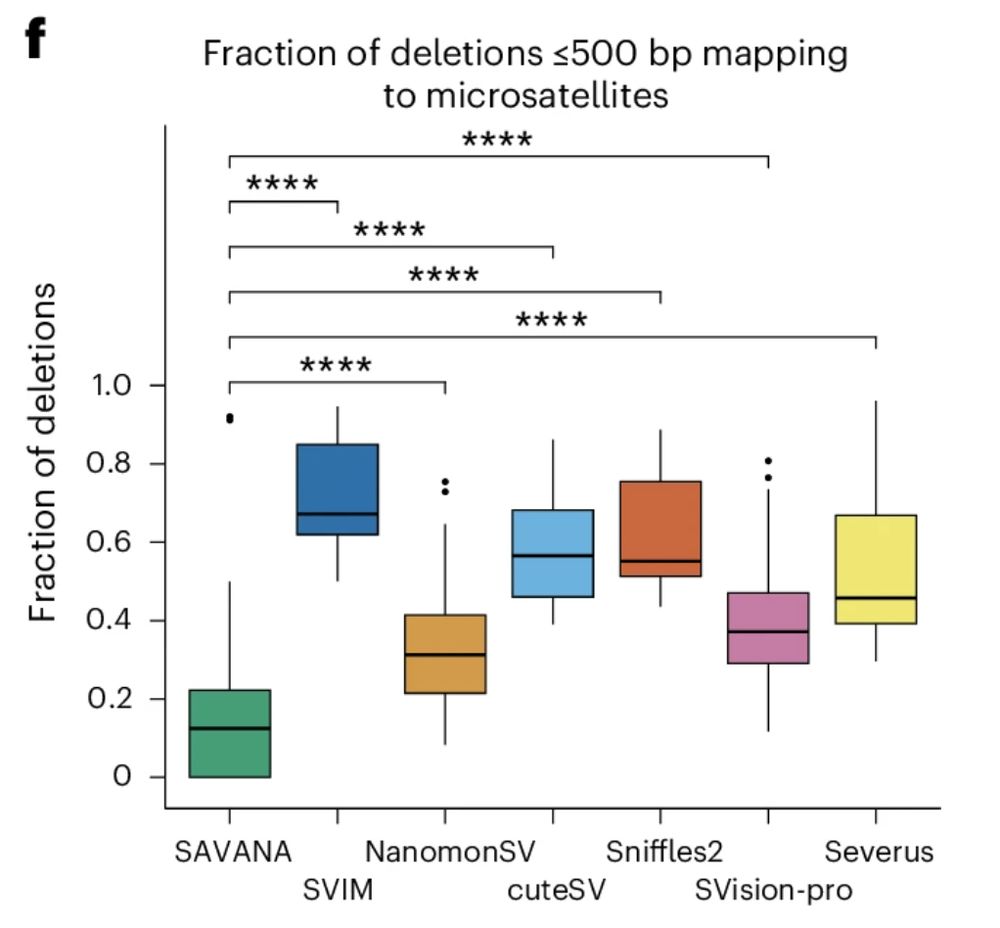
For example, existing methods detect 100s to 1000s of SVs in each sample mapping to microsatellite regions (#SAVANA doesn’t). The tumour types we analysed (sarcomas and glioblastomas) rarely show such levels of repeat instability, which we confirmed for our sample using illumina
May 28, 2025 at 5:36 PM
For example, existing methods detect 100s to 1000s of SVs in each sample mapping to microsatellite regions (#SAVANA doesn’t). The tumour types we analysed (sarcomas and glioblastomas) rarely show such levels of repeat instability, which we confirmed for our sample using illumina
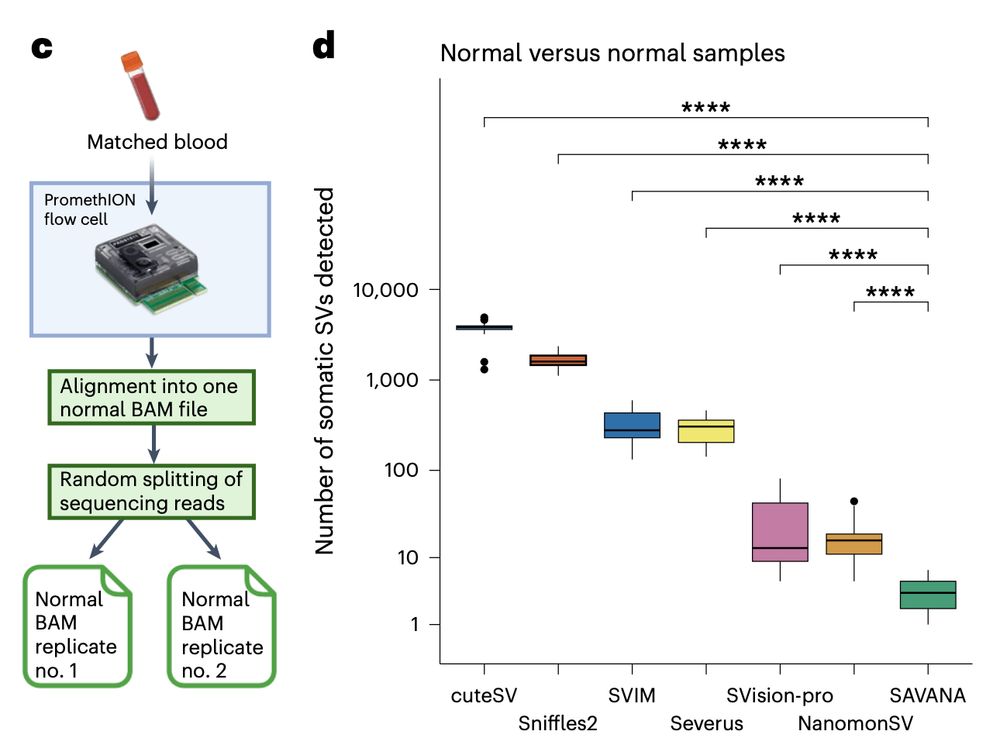
We found the same when using simulated sequencing replicates of the blood samples we use as germline controls. So, what are the false positive SVs called by some algorithms and not by others? What drives such strong differences in performance?
May 28, 2025 at 5:36 PM
We found the same when using simulated sequencing replicates of the blood samples we use as germline controls. So, what are the false positive SVs called by some algorithms and not by others? What drives such strong differences in performance?
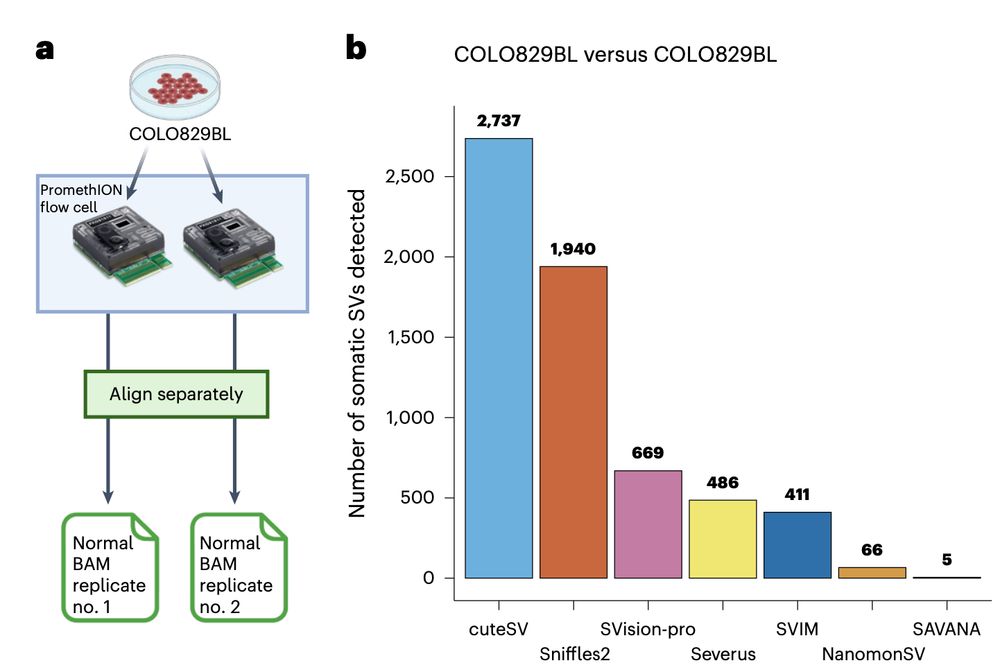
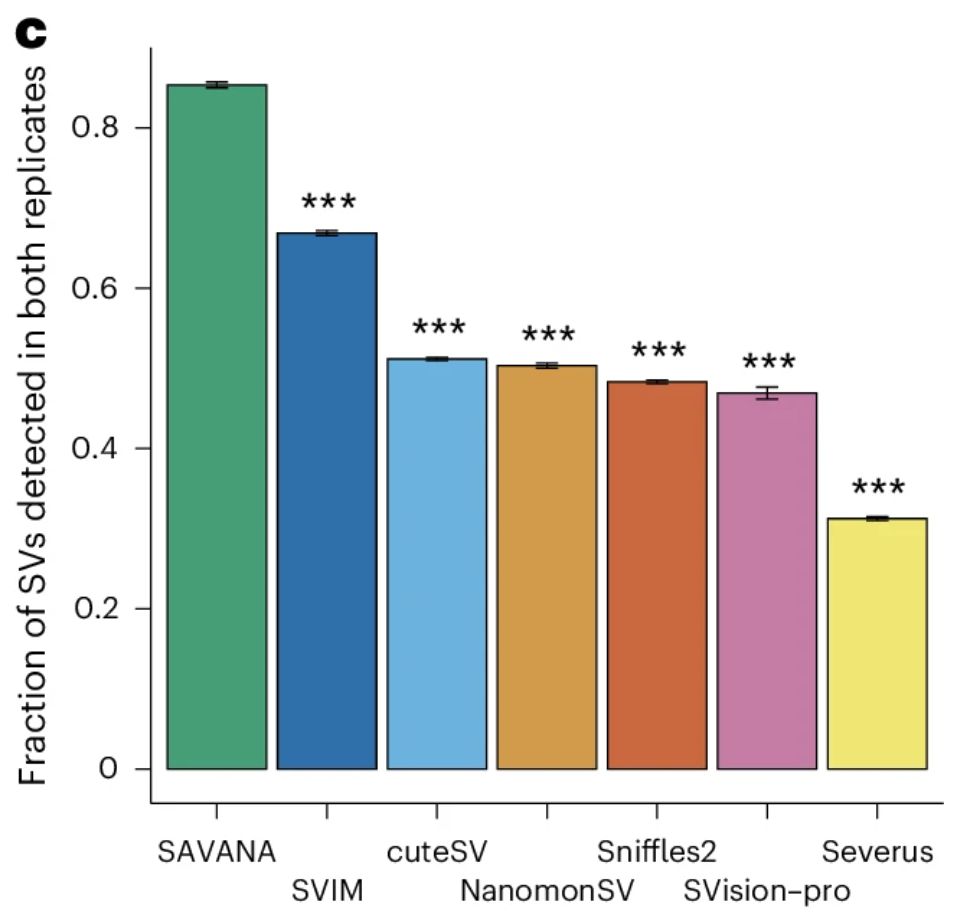
Using sequencing replicates of the normal cell line COLO829BL, we found that SAVANA shows 13- and 82-times higher specificity than the second and third-best performing algorithms (391x higher than the worse performing one). In practice, this means 10s-1000s less false positives..
May 28, 2025 at 5:36 PM
Using sequencing replicates of the normal cell line COLO829BL, we found that SAVANA shows 13- and 82-times higher specificity than the second and third-best performing algorithms (391x higher than the worse performing one). In practice, this means 10s-1000s less false positives..
Well, nullius in verba, let the data speak. SAVANA shows uniform and much higher replication rates across SV clonality levels, sizes, types, samples and genomic regions. Thus, SAVANA's performance if driven by higher sensitivity and specificity!
May 28, 2025 at 5:36 PM
Well, nullius in verba, let the data speak. SAVANA shows uniform and much higher replication rates across SV clonality levels, sizes, types, samples and genomic regions. Thus, SAVANA's performance if driven by higher sensitivity and specificity!
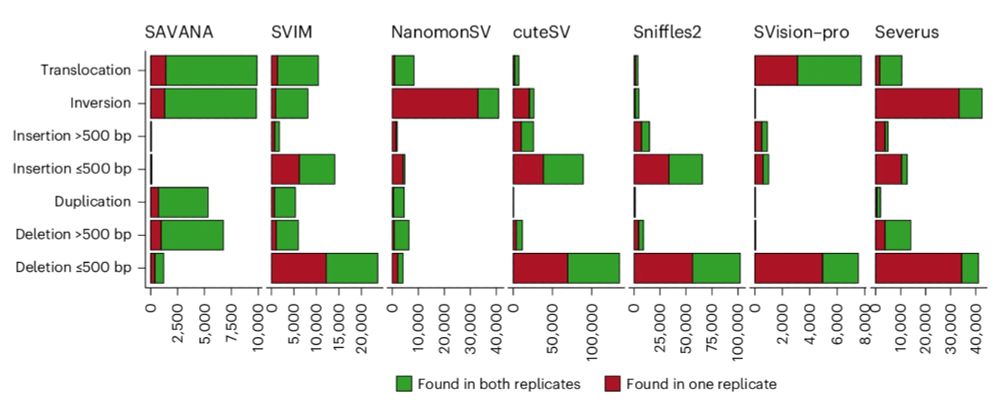
The replication rate of #SAVANA was much higher as compared to existing methods 👇One could argue that lowAF SVs might be missed in one replicate if reads supporting them are assigned to one replicate, and that the many SVs detected by some tools is due to higher sensitivity.
May 28, 2025 at 5:36 PM
The replication rate of #SAVANA was much higher as compared to existing methods 👇One could argue that lowAF SVs might be missed in one replicate if reads supporting them are assigned to one replicate, and that the many SVs detected by some tools is due to higher sensitivity.
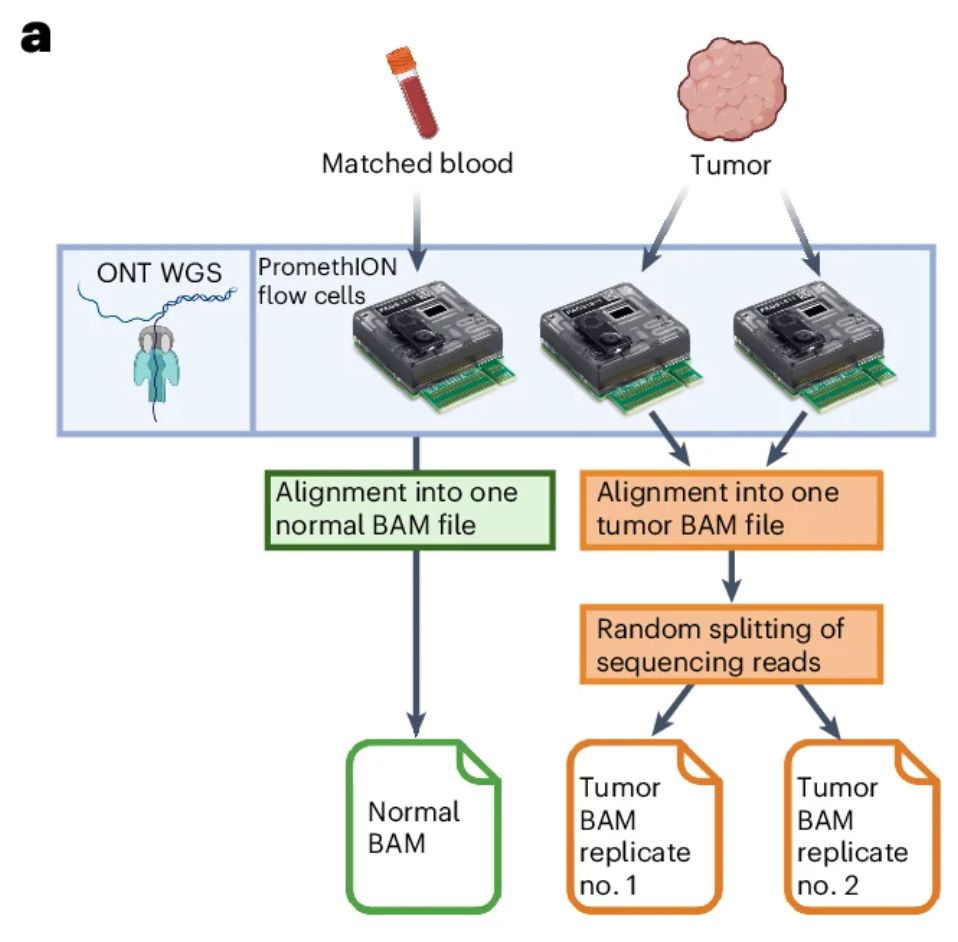
First, we simulated replicates in silico by randomly splitting the sequencing reads from each tumour, reaching even coverage per replicate (note that flowcell yield is variable for ONT). The key idea is: true SVs are detected in both replicates, false positives in just one
May 28, 2025 at 5:36 PM
First, we simulated replicates in silico by randomly splitting the sequencing reads from each tumour, reaching even coverage per replicate (note that flowcell yield is variable for ONT). The key idea is: true SVs are detected in both replicates, false positives in just one
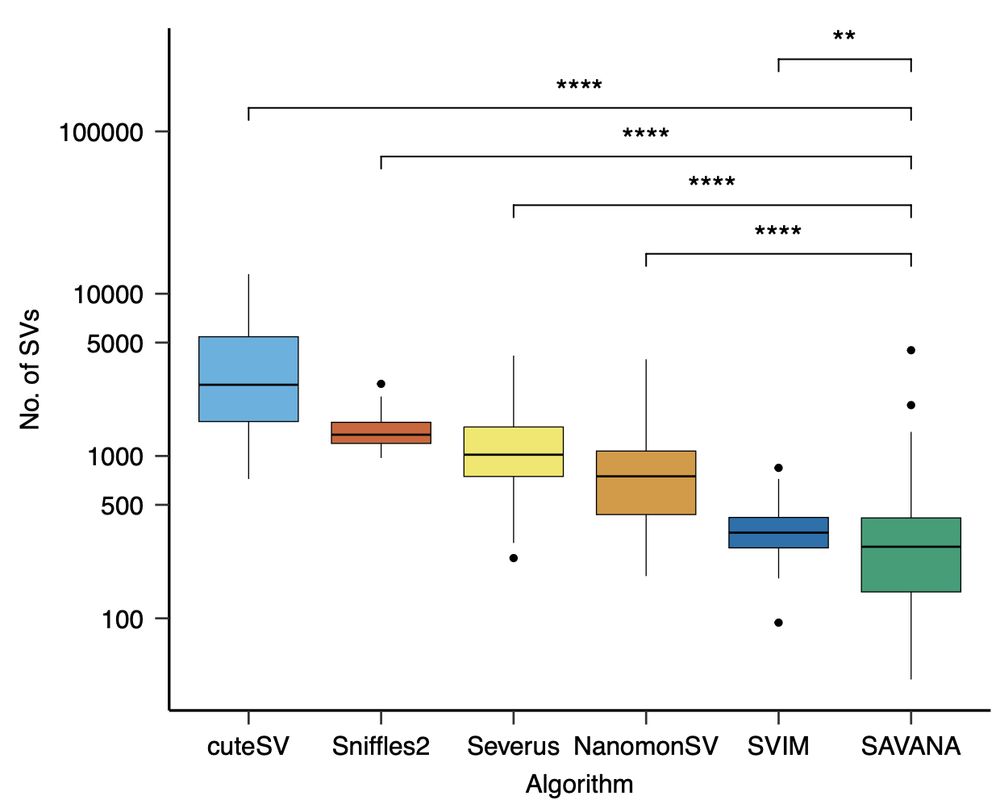
Next, we applied existing SV detection methods designed for germline SV discovery (Sniffles2, cuteSV, SVIM) and for cancer genome analysis (Severus, NanomonSV,SVision-Pro). Sv rates varied across 2 orders of magnitude – so how can we benchmark these tools?
May 28, 2025 at 5:36 PM
Next, we applied existing SV detection methods designed for germline SV discovery (Sniffles2, cuteSV, SVIM) and for cancer genome analysis (Severus, NanomonSV,SVision-Pro). Sv rates varied across 2 orders of magnitude – so how can we benchmark these tools?
Thrilled to see #SAVANA out in @natmethods.nature.com 🥳 SAVANA detects haplotype-resolved somatic SVs, copy number aberrations & infers tumour purity & ploidy using long-read sequencing with or WITHOUT a matched germline control 👇https://www.nature.com/articles/s41592-025-02708-0
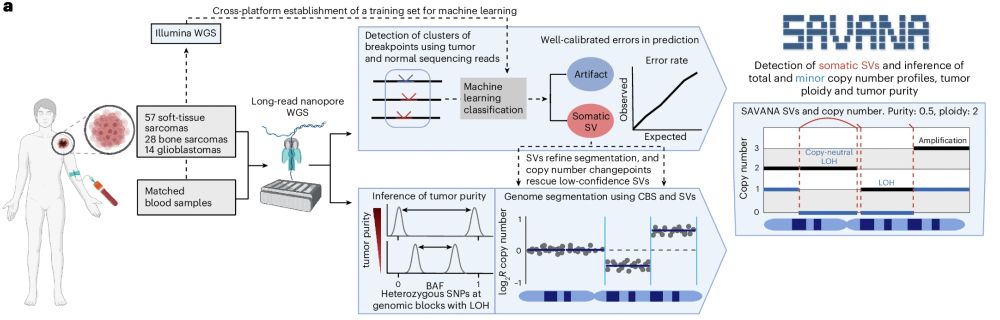
May 28, 2025 at 5:36 PM
Thrilled to see #SAVANA out in @natmethods.nature.com 🥳 SAVANA detects haplotype-resolved somatic SVs, copy number aberrations & infers tumour purity & ploidy using long-read sequencing with or WITHOUT a matched germline control 👇https://www.nature.com/articles/s41592-025-02708-0