




Edu Menezes
@emenezesg.bsky.social
PhD student of computational biology.
Reposted by Edu Menezes

One of the papers they cite in this section discusses some dangers of blindly including data from many different sources (it also has one of the best graphical abstracts ever made) pubs.acs.org/doi/full/10....
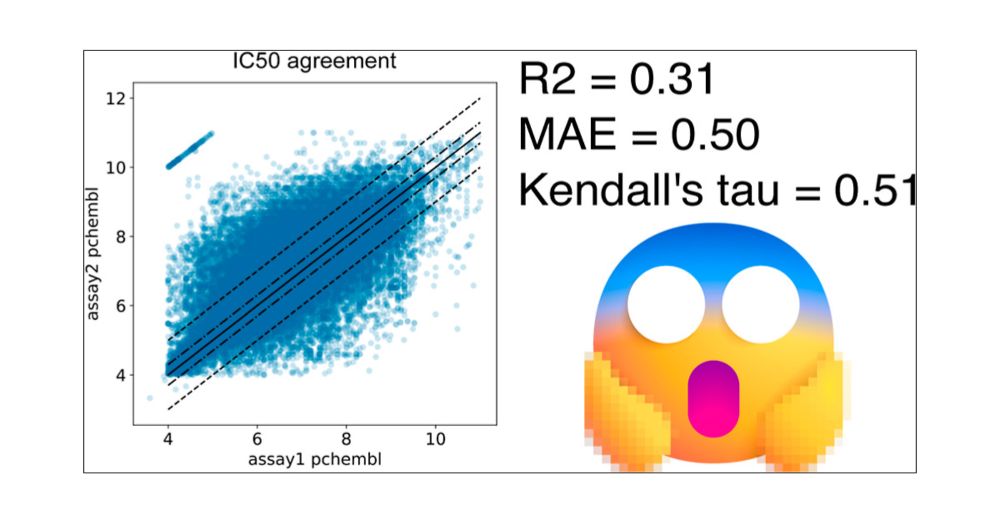
Combining IC50 or Ki Values from Different Sources Is a Source of Significant Noise
As part of the ongoing quest to find or construct large data sets for use in validating new machine learning (ML) approaches for bioactivity prediction, it has become distressingly common for researchers to combine literature IC50 data generated using different assays into a single data set. It is well-known that there are many situations where this is a scientifically risky thing to do, even when the assays are against exactly the same target, but the risks of assays being incompatible are even higher when pulling data from large collections of literature data like ChEMBL. Here, we estimate the amount of noise present in combined data sets using cases where measurements for the same compound are reported in multiple assays against the same target. This approach shows that IC50 assays selected using minimal curation settings have poor agreement with each other: almost 65% of the points differ by more than 0.3 log units, 27% differ by more than one log unit, and the correlation between the assays, as measured by Kendall’s τ, is only 0.51. Requiring that most of the assay metadata in ChEMBL matches (“maximal curation”) in order to combine two assays improves the situation (48% of the points differ by more than 0.3 log units, 13% by more than one log unit, and Kendall’s τ is 0.71) at the expense of having smaller data sets. Surprisingly, our analysis shows similar amounts of noise when combining data from different literature Ki assays. We suggest that good scientific practice requires careful curation when combining data sets from different assays and hope that our maximal curation strategy will help to improve the quality of the data that are being used to build and validate ML models for bioactivity prediction. To help achieve this, the code and ChEMBL queries that we used for the maximal curation approach are available as open-source software in our GitHub repository, https://github.com/rinikerlab/overlapping_assays.
pubs.acs.org
June 7, 2025 at 8:06 AM
One of the papers they cite in this section discusses some dangers of blindly including data from many different sources (it also has one of the best graphical abstracts ever made) pubs.acs.org/doi/full/10....
Reposted by Edu Menezes

MobiDB-lite 4.0: faster prediction of intrinsic protein disorder and structural compactness academic.oup.com/bio...
---
#proteomics #prot-paper
---
#proteomics #prot-paper
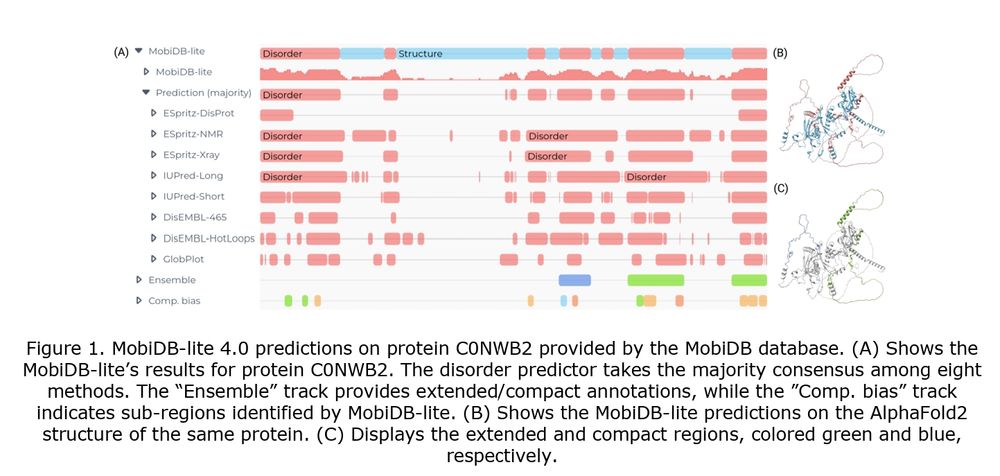
May 12, 2025 at 8:40 AM
MobiDB-lite 4.0: faster prediction of intrinsic protein disorder and structural compactness academic.oup.com/bio...
---
#proteomics #prot-paper
---
#proteomics #prot-paper
Reposted by Edu Menezes
Enhancing Structure-aware Protein Language Models with Efficient Fine-tuning for Various Protein Prediction Tasks www.biorxiv.org/cont...
---
#proteomics #prot-preprint
---
#proteomics #prot-preprint

May 1, 2025 at 12:20 PM
Enhancing Structure-aware Protein Language Models with Efficient Fine-tuning for Various Protein Prediction Tasks www.biorxiv.org/cont...
---
#proteomics #prot-preprint
---
#proteomics #prot-preprint
Reposted by Edu Menezes

Encode protein structures as a series of discrete tokens, train a language model, and sample protein structural conformations given the sequence.
arxiv.org/abs/2410.18403
arxiv.org/abs/2410.18403
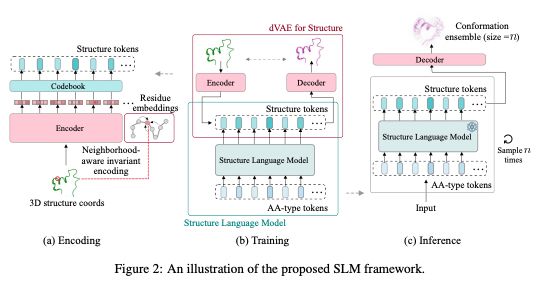
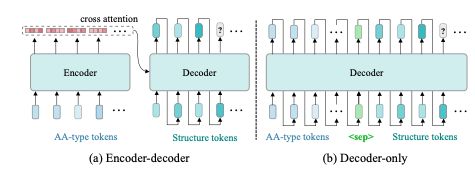
April 25, 2025 at 9:11 PM
Encode protein structures as a series of discrete tokens, train a language model, and sample protein structural conformations given the sequence.
arxiv.org/abs/2410.18403
arxiv.org/abs/2410.18403
Reposted by Edu Menezes

A carioquinha da rede municipal de ensino mandando a mensagem final da matéria do Jornal Nacional sobre o mutirão de vacinação nas escolas públicas, iniciativa do governo federal. Na cidade do Rio toda a comunidade escolar pode ser vacinada, estudantes, professores, funcionários e pais. VIVA O SUS ❤️
April 15, 2025 at 12:00 AM
A carioquinha da rede municipal de ensino mandando a mensagem final da matéria do Jornal Nacional sobre o mutirão de vacinação nas escolas públicas, iniciativa do governo federal. Na cidade do Rio toda a comunidade escolar pode ser vacinada, estudantes, professores, funcionários e pais. VIVA O SUS ❤️
Reposted by Edu Menezes

Big Fantastic Virus Database (BFVD) version 2 improves 31% of predictions through 12 ColabFold recycles. PAEs and MSAs now also available for download and in the webserver.
🌐https://bfvd.foldseek.com
💾https://bfvd.steineggerlab.workers.dev/
1/3
🌐https://bfvd.foldseek.com
💾https://bfvd.steineggerlab.workers.dev/
1/3
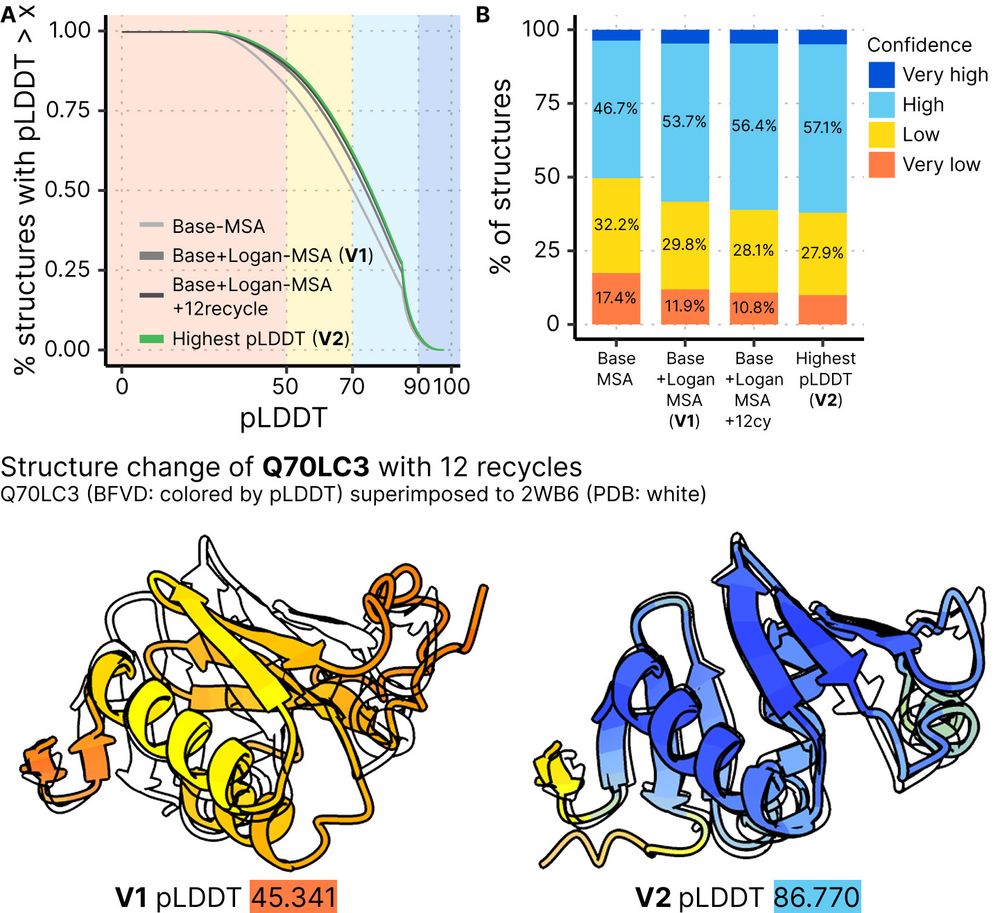
March 31, 2025 at 5:07 AM
Big Fantastic Virus Database (BFVD) version 2 improves 31% of predictions through 12 ColabFold recycles. PAEs and MSAs now also available for download and in the webserver.
🌐https://bfvd.foldseek.com
💾https://bfvd.steineggerlab.workers.dev/
1/3
🌐https://bfvd.foldseek.com
💾https://bfvd.steineggerlab.workers.dev/
1/3
Reposted by Edu Menezes
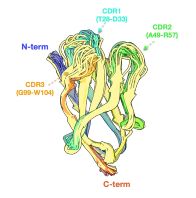
A classifier that distinguishes between flexible and rigid CDR3 loops (rigid loops are more desirable as they bind with greater affinity and specificity than flexible loops) www.biorxiv.org/content/10.1...

March 20, 2025 at 6:53 AM
A classifier that distinguishes between flexible and rigid CDR3 loops (rigid loops are more desirable as they bind with greater affinity and specificity than flexible loops) www.biorxiv.org/content/10.1...