



Anthony Gitter
@anthonygitter.bsky.social
Computational biologist; Associate Prof. at University of Wisconsin-Madison; Jeanne M. Rowe Chair at Morgridge Institute
Looks very interesting. Can I think of this like a more extreme form of the evotuning from UniRep or doi.org/10.1101/2024... except it uses one sequence instead of the sequence plus homologs?

Protein Language Model Fitness Is a Matter of Preference
Leveraging billions of years of evolution, scientists have trained protein language models (pLMs) to understand the sequence and structure space of proteins aiding in the design of more functional pro...
doi.org
October 23, 2025 at 10:23 PM
Looks very interesting. Can I think of this like a more extreme form of the evotuning from UniRep or doi.org/10.1101/2024... except it uses one sequence instead of the sequence plus homologs?
Bioconductor R package: bioconductor.org/packages/MPAC
Shiny app to explore results in manuscript: connect.doit.wisc.edu/content/122/
Shiny app to explore results in manuscript: connect.doit.wisc.edu/content/122/

MPAC
Multi-omic Pathway Analysis of Cells (MPAC), integrates multi-omic data for understanding cellular mechanisms. It predicts novel patient groups with distinct pathway profiles as well as identifying ke...
bioconductor.org
October 10, 2025 at 2:56 PM
Bioconductor R package: bioconductor.org/packages/MPAC
Shiny app to explore results in manuscript: connect.doit.wisc.edu/content/122/
Shiny app to explore results in manuscript: connect.doit.wisc.edu/content/122/
MPAC uses PARADIGM as the probabilistic model but makes many improvements:
- data-driven omic data discretization
- permutation testing to eliminate spurious predictions
- full workflow and downstream analyses in an R package
- Shiny app for interactive visualization
- data-driven omic data discretization
- permutation testing to eliminate spurious predictions
- full workflow and downstream analyses in an R package
- Shiny app for interactive visualization
October 10, 2025 at 2:56 PM
MPAC uses PARADIGM as the probabilistic model but makes many improvements:
- data-driven omic data discretization
- permutation testing to eliminate spurious predictions
- full workflow and downstream analyses in an R package
- Shiny app for interactive visualization
- data-driven omic data discretization
- permutation testing to eliminate spurious predictions
- full workflow and downstream analyses in an R package
- Shiny app for interactive visualization
I found out that Neurosnap offers ESMFold via API neurosnap.ai/service/ESMF...
I may test how many calls are possible with the free academic plan to see if it is worthwhile to update my repo.
I may test how many calls are possible with the free academic plan to see if it is worthwhile to update my repo.

🧬 Use ESMFold Online | Neurosnap
Bulk protein structure prediction model that only requires a single amino acid sequence as input. Much faster than AlphaFold2 since no MSAs are required (but slightly less accurate too).
neurosnap.ai
October 9, 2025 at 2:25 AM
I found out that Neurosnap offers ESMFold via API neurosnap.ai/service/ESMF...
I may test how many calls are possible with the free academic plan to see if it is worthwhile to update my repo.
I may test how many calls are possible with the free academic plan to see if it is worthwhile to update my repo.
The main GitHub repo github.com/gitter-lab/m... links to the extensive resources for running Rosetta simulations at scale to generate new training data, training METL models, running our models, and accessing our datasets. 8/

GitHub - gitter-lab/metl: Mutational Effect Transfer Learning (METL) framework for pretraining and finetuning biophysics-informed protein language models
Mutational Effect Transfer Learning (METL) framework for pretraining and finetuning biophysics-informed protein language models - gitter-lab/metl
github.com
September 11, 2025 at 5:00 PM
The main GitHub repo github.com/gitter-lab/m... links to the extensive resources for running Rosetta simulations at scale to generate new training data, training METL models, running our models, and accessing our datasets. 8/
We can use METL for low-N protein design. We trained METL on Rosetta simulations of GFP biophysical attributes and only 64 experimental examples of GFP brightness. It designed fluorescent 5 and 10 mutants, including some with mutants entirely outside training set mutations. 7/

September 11, 2025 at 5:00 PM
We can use METL for low-N protein design. We trained METL on Rosetta simulations of GFP biophysical attributes and only 64 experimental examples of GFP brightness. It designed fluorescent 5 and 10 mutants, including some with mutants entirely outside training set mutations. 7/
A powerful aspect of pretraining on biophysical simulations is that the simulations can be customized to match the protein function and experimental assay. Our expanded simulations of the GB1-IgG complex with Rosetta InterfaceAnalyzer improve METL predictions of GB1 binding. 6/

September 11, 2025 at 5:00 PM
A powerful aspect of pretraining on biophysical simulations is that the simulations can be customized to match the protein function and experimental assay. Our expanded simulations of the GB1-IgG complex with Rosetta InterfaceAnalyzer improve METL predictions of GB1 binding. 6/
We also benchmark METL on four types of difficult extrapolation. For instance, positional extrapolation provides training data from some sequence positions and tests predictions at different sequence positions. Linear regression completely fails in this setting. 5/

September 11, 2025 at 5:00 PM
We also benchmark METL on four types of difficult extrapolation. For instance, positional extrapolation provides training data from some sequence positions and tests predictions at different sequence positions. Linear regression completely fails in this setting. 5/
We compare these approaches on deep mutational scanning datasets with increasing training set sizes. Biophysical pretraining helps METL generalize well with small training sets. However, augmented linear regression with EVE scores is great on some of these assays. 4/

September 11, 2025 at 5:00 PM
We compare these approaches on deep mutational scanning datasets with increasing training set sizes. Biophysical pretraining helps METL generalize well with small training sets. However, augmented linear regression with EVE scores is great on some of these assays. 4/
METL models pretrained on Rosetta biophysical attributes learn different protein representations than general protein language models like ESM-2 or protein family-specific models like EVE. These new representations are valuable for machine learning-guided protein engineering. 3/
September 11, 2025 at 5:00 PM
METL models pretrained on Rosetta biophysical attributes learn different protein representations than general protein language models like ESM-2 or protein family-specific models like EVE. These new representations are valuable for machine learning-guided protein engineering. 3/
Most protein language models train on natural protein sequence data and use the underlying evolutionary signals to score sequence variants. Instead, METL trains on @rosettacommons.bsky.social data, learning from simulated biophyiscal attributes of the sequence variants we select. 2/
September 11, 2025 at 5:00 PM
Most protein language models train on natural protein sequence data and use the underlying evolutionary signals to score sequence variants. Instead, METL trains on @rosettacommons.bsky.social data, learning from simulated biophyiscal attributes of the sequence variants we select. 2/
Paper: arxiv.org/abs/2507.12574
GitHub: github.com/gitter-lab/A...
Datasets: doi.org/10.5281/zeno...
7/
GitHub: github.com/gitter-lab/A...
Datasets: doi.org/10.5281/zeno...
7/

Assay2Mol: large language model-based drug design using BioAssay context
Scientific databases aggregate vast amounts of quantitative data alongside descriptive text. In biochemistry, molecule screening assays evaluate the functional responses of candidate molecules against...
arxiv.org
July 18, 2025 at 3:13 PM
Paper: arxiv.org/abs/2507.12574
GitHub: github.com/gitter-lab/A...
Datasets: doi.org/10.5281/zeno...
7/
GitHub: github.com/gitter-lab/A...
Datasets: doi.org/10.5281/zeno...
7/
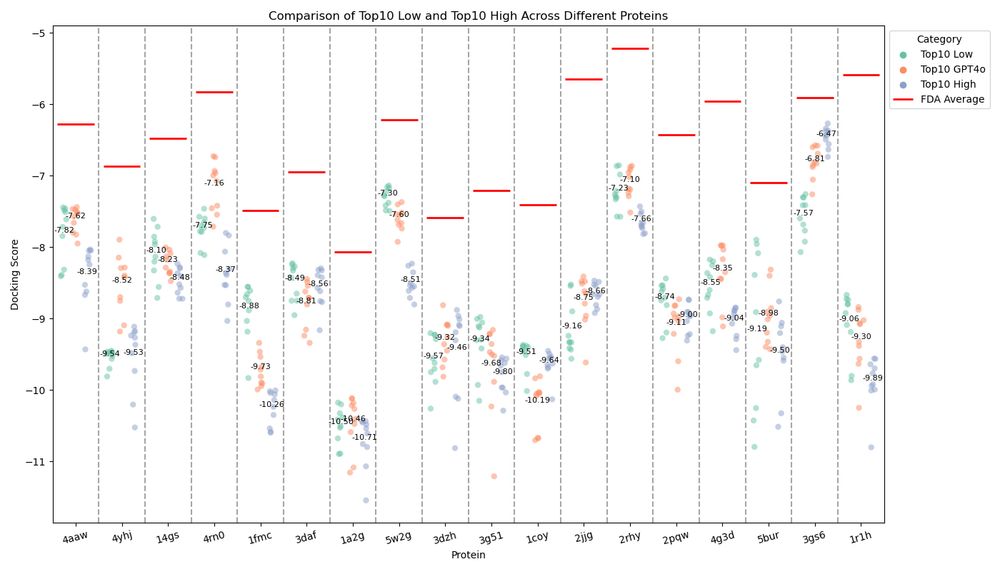
There are many more results and controls in the paper. Here's how the best (most negative) docking scores change when we use relevant assays, irrelevant assays, or no assays as context for generation with GPT-4o. In the majority of cases, but not all, relevant context helps. 6/
July 18, 2025 at 3:13 PM
There are many more results and controls in the paper. Here's how the best (most negative) docking scores change when we use relevant assays, irrelevant assays, or no assays as context for generation with GPT-4o. In the majority of cases, but not all, relevant context helps. 6/
This generally has the desired effects across multiple LLMs and queried protein targets, with the caveat that our core results are based on AutoDock Vina scores. Assessing generated molecules with docking is admittedly frustrating. 5/
July 18, 2025 at 3:13 PM
This generally has the desired effects across multiple LLMs and queried protein targets, with the caveat that our core results are based on AutoDock Vina scores. Assessing generated molecules with docking is admittedly frustrating. 5/
We embed the BioAssay data into a vectorbase, retrieve initial candidate assays, and do further LLM-based filtering and summarization. We select some active and inactive molecules from the BioAssay data table. This is all used for in-context learning and molecule generation. 4/
July 18, 2025 at 3:13 PM
We embed the BioAssay data into a vectorbase, retrieve initial candidate assays, and do further LLM-based filtering and summarization. We select some active and inactive molecules from the BioAssay data table. This is all used for in-context learning and molecule generation. 4/
A proof of concept study from our collaborators showed that mining this PubChem data successfully identified new candidates for a target phenotype, oxidative phosphorylation doi.org/10.1186/s133....
We wanted to generalize that for any new query and assess the effectiveness. 3/
We wanted to generalize that for any new query and assess the effectiveness. 3/
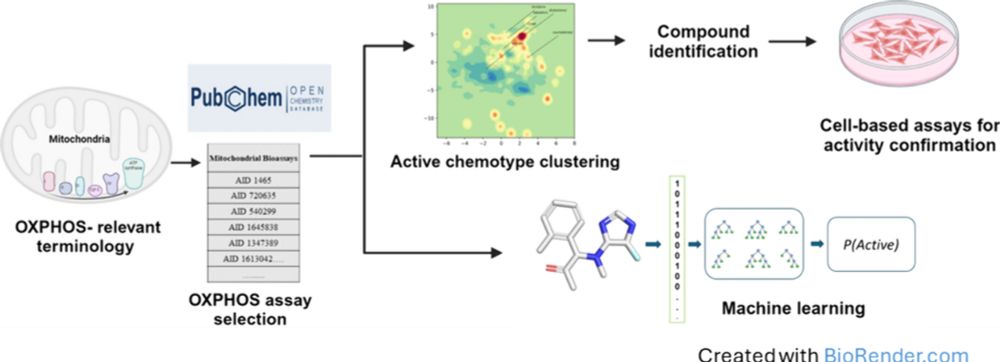
Data mining of PubChem bioassay records reveals diverse OXPHOS inhibitory chemotypes as potential therapeutic agents against ovarian cancer - Journal of Cheminformatics
Focused screening on target-prioritized compound sets can be an efficient alternative to high throughput screening (HTS). For most biomolecular targets, compound prioritization models depend on prior ...
doi.org
July 18, 2025 at 3:13 PM
A proof of concept study from our collaborators showed that mining this PubChem data successfully identified new candidates for a target phenotype, oxidative phosphorylation doi.org/10.1186/s133....
We wanted to generalize that for any new query and assess the effectiveness. 3/
We wanted to generalize that for any new query and assess the effectiveness. 3/
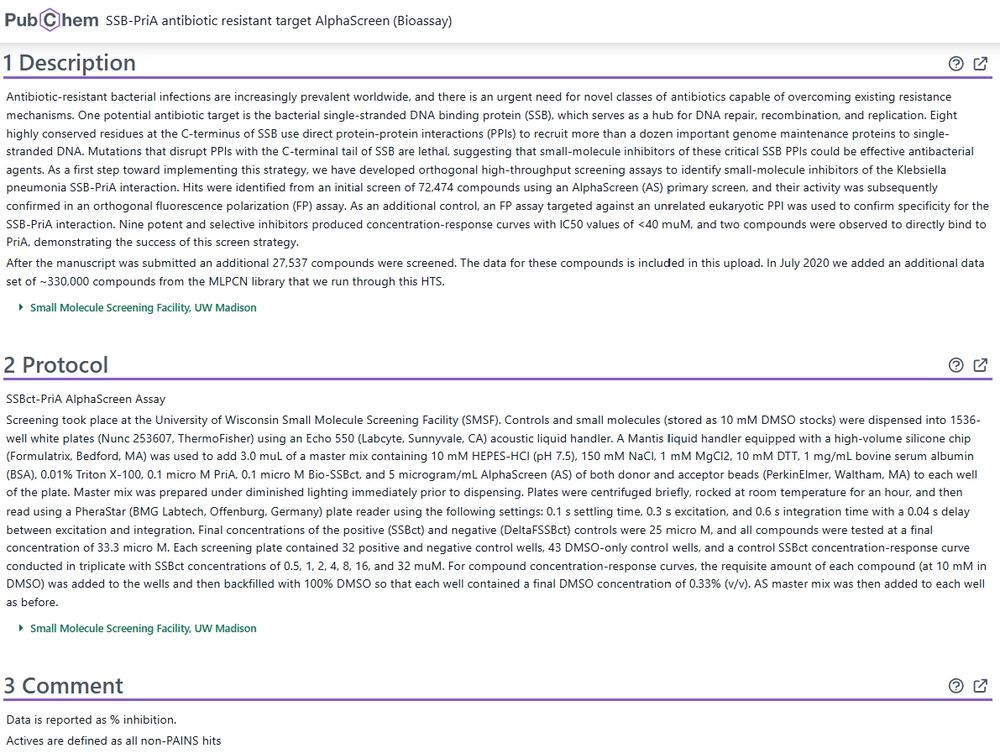
PubChem BioAssays can contain a lot of information about why and how an assay was run. Here's an example from our collaborators. pubchem.ncbi.nlm.nih.gov/bioassay/127...
There are now 1.7M PubChem BioAssays ranging in scale from a few tested molecules to high-throughput screens. 2/
There are now 1.7M PubChem BioAssays ranging in scale from a few tested molecules to high-throughput screens. 2/
July 18, 2025 at 3:13 PM
PubChem BioAssays can contain a lot of information about why and how an assay was run. Here's an example from our collaborators. pubchem.ncbi.nlm.nih.gov/bioassay/127...
There are now 1.7M PubChem BioAssays ranging in scale from a few tested molecules to high-throughput screens. 2/
There are now 1.7M PubChem BioAssays ranging in scale from a few tested molecules to high-throughput screens. 2/