



Juan Viguera Diez
@viguera10.bsky.social
PhD student at AstraZeneca and Chalmers University of Technology. Machine Learning for natural sciences (Drug discovery).
Reposted by Juan Viguera Diez

New preprint out!
We present "Transferable Generative Models Bridge Femtosecond to Nanosecond Time-Step Molecular Dynamics,"
We present "Transferable Generative Models Bridge Femtosecond to Nanosecond Time-Step Molecular Dynamics,"

October 10, 2025 at 1:45 PM
New preprint out!
We present "Transferable Generative Models Bridge Femtosecond to Nanosecond Time-Step Molecular Dynamics,"
We present "Transferable Generative Models Bridge Femtosecond to Nanosecond Time-Step Molecular Dynamics,"
Reposted by Juan Viguera Diez

Excited to share that our paper "LAGOM: A transformer-based chemical language model for drug metabolite prediction" has been accepted in AILSCI!
doi.org/10.1016/j.ai...
Work led by Sofia Larsson and Miranda Carlsson, with @rbeckmann.bsky.social and Filip Miljković (AZ)!
#compchem #chemsky
doi.org/10.1016/j.ai...
Work led by Sofia Larsson and Miranda Carlsson, with @rbeckmann.bsky.social and Filip Miljković (AZ)!
#compchem #chemsky

September 22, 2025 at 2:06 PM
Excited to share that our paper "LAGOM: A transformer-based chemical language model for drug metabolite prediction" has been accepted in AILSCI!
doi.org/10.1016/j.ai...
Work led by Sofia Larsson and Miranda Carlsson, with @rbeckmann.bsky.social and Filip Miljković (AZ)!
#compchem #chemsky
doi.org/10.1016/j.ai...
Work led by Sofia Larsson and Miranda Carlsson, with @rbeckmann.bsky.social and Filip Miljković (AZ)!
#compchem #chemsky
Reposted by Juan Viguera Diez
The official opening to this position was just posted: www.chalmers.se/en/about-cha...
June 10, 2025 at 1:48 PM
The official opening to this position was just posted: www.chalmers.se/en/about-cha...
Reposted by Juan Viguera Diez
Registration for this years CHAIR Structured Learning Workshop is open. Speakers include: Klaus Robert Müller, Jens Sjölund, @alextong.bsky.social ,
@janstuehmer.bsky.social, @arnauddoucet.bsky.social, @marcocuturi.bsky.social , Marta Betcke,
Elena Agliari, Beatriz Seoane, Alessandro Ingrosso
@janstuehmer.bsky.social, @arnauddoucet.bsky.social, @marcocuturi.bsky.social , Marta Betcke,
Elena Agliari, Beatriz Seoane, Alessandro Ingrosso

2025 CHAIR Structured Learning Workshop
Welcome to the 2025 Chalmers AI Research Center Workshop for Structured Learning. In this workshop we broadly cover topics related to Structured Learning targeting specifically the following questions...
ui.ungpd.com
April 24, 2025 at 1:48 PM
Registration for this years CHAIR Structured Learning Workshop is open. Speakers include: Klaus Robert Müller, Jens Sjölund, @alextong.bsky.social ,
@janstuehmer.bsky.social, @arnauddoucet.bsky.social, @marcocuturi.bsky.social , Marta Betcke,
Elena Agliari, Beatriz Seoane, Alessandro Ingrosso
@janstuehmer.bsky.social, @arnauddoucet.bsky.social, @marcocuturi.bsky.social , Marta Betcke,
Elena Agliari, Beatriz Seoane, Alessandro Ingrosso
Reposted by Juan Viguera Diez
Join us in Gothenburg for the 3rd CHAIR Structured Learning Workshop! Very exciting line-up of speakers and we expect lots of engaging discussions, too. Attendance is free but you need to apply as we have limited slots.
Registration for this years CHAIR Structured Learning Workshop is open. Speakers include: Klaus Robert Müller, Jens Sjölund, @alextong.bsky.social ,
@janstuehmer.bsky.social, @arnauddoucet.bsky.social, @marcocuturi.bsky.social , Marta Betcke,
Elena Agliari, Beatriz Seoane, Alessandro Ingrosso
@janstuehmer.bsky.social, @arnauddoucet.bsky.social, @marcocuturi.bsky.social , Marta Betcke,
Elena Agliari, Beatriz Seoane, Alessandro Ingrosso
2025 CHAIR Structured Learning Workshop
Welcome to the 2025 Chalmers AI Research Center Workshop for Structured Learning. In this workshop we broadly cover topics related to Structured Learning targeting specifically the following questions...
ui.ungpd.com
April 24, 2025 at 2:58 PM
Join us in Gothenburg for the 3rd CHAIR Structured Learning Workshop! Very exciting line-up of speakers and we expect lots of engaging discussions, too. Attendance is free but you need to apply as we have limited slots.
Excited to present our poster on Boltzmann priors for Implicit Transfer Operators tomorrow at @iclr-conf.bsky.social!
See you tomorrow at poster 13, 10-12:30.
See you tomorrow at poster 13, 10-12:30.

April 24, 2025 at 8:20 AM
Excited to present our poster on Boltzmann priors for Implicit Transfer Operators tomorrow at @iclr-conf.bsky.social!
See you tomorrow at poster 13, 10-12:30.
See you tomorrow at poster 13, 10-12:30.
Reposted by Juan Viguera Diez
The Chalmers AI4Science speakers for the spring term have just been announced please check the homepage for all the details: psolsson.github.io/AI4ScienceSe...
AI and Machine Learning in the Natural Sciences | AI4ScienceSeminar
A simple, whitespace theme for academics. Based on [*folio](https://github.com/bogoli/-folio) design.
psolsson.github.io
January 24, 2025 at 11:00 PM
The Chalmers AI4Science speakers for the spring term have just been announced please check the homepage for all the details: psolsson.github.io/AI4ScienceSe...
Reposted by Juan Viguera Diez

Check out our new paper!
Protein assembly in membranes is crucial yet elusive. Why steer when you can just observe? We introduce a bias-free simulation method that captures the full picture of transmembrane dimerization—free energies, mechanisms, and rates!
pubs.acs.org/doi/10.1021/...
Protein assembly in membranes is crucial yet elusive. Why steer when you can just observe? We introduce a bias-free simulation method that captures the full picture of transmembrane dimerization—free energies, mechanisms, and rates!
pubs.acs.org/doi/10.1021/...
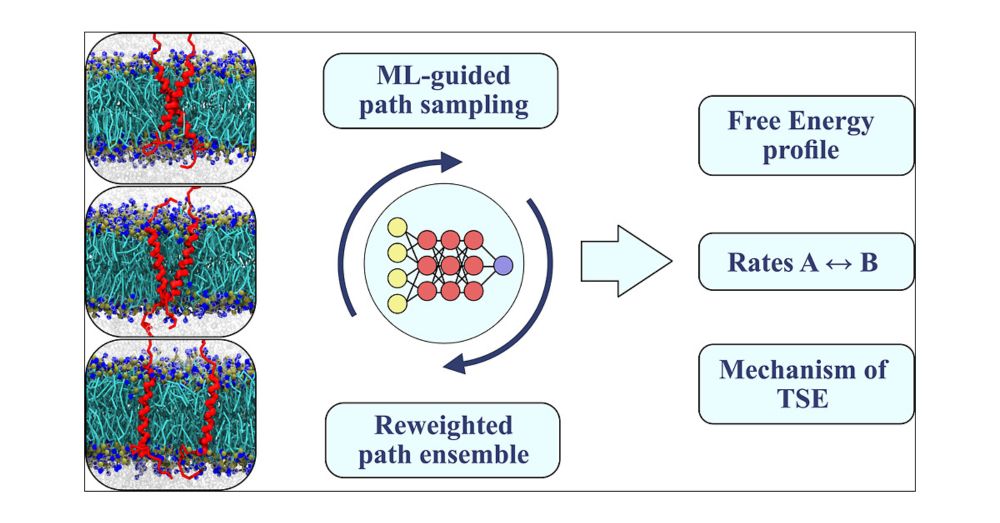
Free Energy, Rates, and Mechanism of Transmembrane Dimerization in Lipid Bilayers from Dynamically Unbiased Molecular Dynamics Simulations
The assembly of proteins in membranes plays a key role in many crucial cellular pathways. Despite their importance, characterizing transmembrane assembly remains challenging for experiments and simula...
pubs.acs.org
January 24, 2025 at 2:37 PM
Check out our new paper!
Protein assembly in membranes is crucial yet elusive. Why steer when you can just observe? We introduce a bias-free simulation method that captures the full picture of transmembrane dimerization—free energies, mechanisms, and rates!
pubs.acs.org/doi/10.1021/...
Protein assembly in membranes is crucial yet elusive. Why steer when you can just observe? We introduce a bias-free simulation method that captures the full picture of transmembrane dimerization—free energies, mechanisms, and rates!
pubs.acs.org/doi/10.1021/...
Reposted by Juan Viguera Diez
1/1 Papers accepted at ICLR2025 Congrats @viguera10.bsky.social and 1/1 papers accepted at AISTATS2025 congrats @rossirwin.bsky.social
January 22, 2025 at 5:32 PM
1/1 Papers accepted at ICLR2025 Congrats @viguera10.bsky.social and 1/1 papers accepted at AISTATS2025 congrats @rossirwin.bsky.social
Reposted by Juan Viguera Diez
Come join us on an exciting joint PhD project on ML for protein function with @kailalab.bsky.social -- We are looking for a strong quantitative candidate with prior experience in ML. The position is based in Gothenburg and is fully funded for 5 years.
www.chalmers.se/en/about-cha...
Vacancies
www.chalmers.se
December 23, 2024 at 8:17 AM
Come join us on an exciting joint PhD project on ML for protein function with @kailalab.bsky.social -- We are looking for a strong quantitative candidate with prior experience in ML. The position is based in Gothenburg and is fully funded for 5 years.
www.chalmers.se/en/about-cha...
Reposted by Juan Viguera Diez

I am hiring a postdoctoral scholar with a start date summer or fall 2025. Projects will be focused on thermodynamically consistent generative models, broadly defined. If you’re interested, please send a CV and one paragraph about why you think you’d be a good fit to rotskoff@stanford.edu
December 23, 2024 at 5:31 PM
I am hiring a postdoctoral scholar with a start date summer or fall 2025. Projects will be focused on thermodynamically consistent generative models, broadly defined. If you’re interested, please send a CV and one paragraph about why you think you’d be a good fit to rotskoff@stanford.edu
Reposted by Juan Viguera Diez

AMARO: All Heavy-Atom Transferable Neural Network Potentials of Protein Thermodynamics
A novel machine-learning-based force field for protein thermodynamics. It uses an all-heavy-atom approach without hydrogens to simplify simulations while maintaining key dynamics.
pubs.acs.org/doi/10.1021/...
A novel machine-learning-based force field for protein thermodynamics. It uses an all-heavy-atom approach without hydrogens to simplify simulations while maintaining key dynamics.
pubs.acs.org/doi/10.1021/...

November 19, 2024 at 3:16 PM
AMARO: All Heavy-Atom Transferable Neural Network Potentials of Protein Thermodynamics
A novel machine-learning-based force field for protein thermodynamics. It uses an all-heavy-atom approach without hydrogens to simplify simulations while maintaining key dynamics.
pubs.acs.org/doi/10.1021/...
A novel machine-learning-based force field for protein thermodynamics. It uses an all-heavy-atom approach without hydrogens to simplify simulations while maintaining key dynamics.
pubs.acs.org/doi/10.1021/...
Reposted by Juan Viguera Diez

Thrilled to announce Boltz-1, the first open-source and commercially available model to achieve AlphaFold3-level accuracy on biomolecular structure prediction! An exciting collaboration with Jeremy, Saro, and an amazing team at MIT and Genesis Therapeutics. A thread!
November 17, 2024 at 4:20 PM
Thrilled to announce Boltz-1, the first open-source and commercially available model to achieve AlphaFold3-level accuracy on biomolecular structure prediction! An exciting collaboration with Jeremy, Saro, and an amazing team at MIT and Genesis Therapeutics. A thread!
Reposted by Juan Viguera Diez
Chemists use NMR spectroscopy to identify molecules, but interpreting spectra is laborious and error prone. We show the process can be automated end-to-end using a well-designed Molecular GPT. Importantly, we also make predictions of substructures for interpretability. pubs.acs.org/doi/10.1021/...
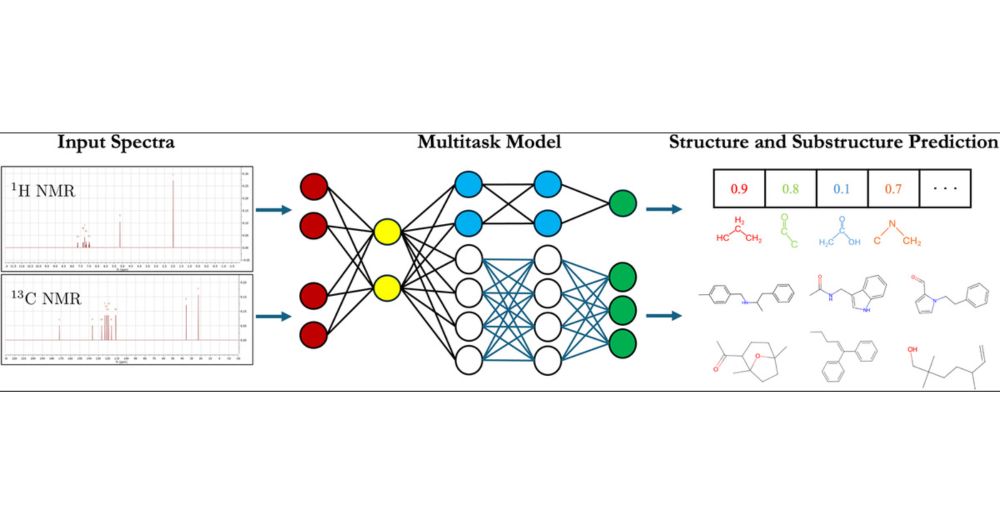
Accurate and Efficient Structure Elucidation from Routine One-Dimensional NMR Spectra Using Multitask Machine Learning
Rapid determination of molecular structures can greatly accelerate workflows across many chemical disciplines. However, elucidating structure using only one-dimensional (1D) NMR spectra, the most readily accessible data, remains an extremely challenging problem because of the combinatorial explosion of the number of possible molecules as the number of constituent atoms is increased. Here, we introduce a multitask machine learning framework that predicts the molecular structure (formula and connectivity) of an unknown compound solely based on its 1D 1H and/or 13C NMR spectra. First, we show how a transformer architecture can be constructed to efficiently solve the task, traditionally performed by chemists, of assembling large numbers of molecular fragments into molecular structures. Integrating this capability with a convolutional neural network, we build an end-to-end model for predicting structure from spectra that is fast and accurate. We demonstrate the effectiveness of this framework on molecules with up to 19 heavy (non-hydrogen) atoms, a size for which there are trillions of possible structures. Without relying on any prior chemical knowledge such as the molecular formula, we show that our approach predicts the exact molecule 69.6% of the time within the first 15 predictions, reducing the search space by up to 11 orders of magnitude.
pubs.acs.org
November 13, 2024 at 6:22 PM
Chemists use NMR spectroscopy to identify molecules, but interpreting spectra is laborious and error prone. We show the process can be automated end-to-end using a well-designed Molecular GPT. Importantly, we also make predictions of substructures for interpretability. pubs.acs.org/doi/10.1021/...