



Moez Dawood
@moezdawood.bsky.social
Pinned
Moez Dawood
@moezdawood.bsky.social
· Mar 21
🚨 Big news at #ACMG2025! 🚨
Today we’re announcing global democratization of deidentified allele count + frequency data with population breakdown from the first ~250k short-read WGS in All of Us designed to plug straight into clinical workflows. It is ~1.1 billion unique variants! 🧬💡
🧵 (1/4)
Today we’re announcing global democratization of deidentified allele count + frequency data with population breakdown from the first ~250k short-read WGS in All of Us designed to plug straight into clinical workflows. It is ~1.1 billion unique variants! 🧬💡
🧵 (1/4)
Reposted by Moez Dawood

Learn about the major accomplishments of @gregor-research.bsky.social. R. Gibbs, @bcmhgsc.bsky.social @moezdawood.bsky.social #LupskiLab @sedlazeck.bsky.social @poseypod.bsky.social @bcmhouston.bsky.social S. Montgomery @stanfordmedicine.bsky.social @nature.com blogs.bcm.edu/2025/11/18/f...

How GREGoR Consortium is advancing the diagnostics of rare diseases
Learn about the major accomplishments of the consortium’s first five years and the frontiers in genomic medicine that researchers will tackle next.
blogs.bcm.edu
November 18, 2025 at 9:04 PM
Learn about the major accomplishments of @gregor-research.bsky.social. R. Gibbs, @bcmhgsc.bsky.social @moezdawood.bsky.social #LupskiLab @sedlazeck.bsky.social @poseypod.bsky.social @bcmhouston.bsky.social S. Montgomery @stanfordmedicine.bsky.social @nature.com blogs.bcm.edu/2025/11/18/f...
Reposted by Moez Dawood

@moezdawood.bsky.social created the AllofUs 250k annotator for OpenCRAVAT to improve genomic research by integrating diverse data, including ancestry-specific allele frequencies. You can read more about Moez's experience developing the annotator here: www.opencravat.org/bringing-gen...
#opensource
#opensource

Bringing Genetic Diversity to the Forefront: Moez Dawood on the AllofUs 250k Annotator for OpenCRAVAT
Moez Dawood, an MD/PhD student at Baylor College of Medicine, has recently developed the AllofUs 250k annotator for OpenCRAVAT, a genomic annotation platform designed to handle both single variant…
www.opencravat.org
August 15, 2025 at 4:14 PM
@moezdawood.bsky.social created the AllofUs 250k annotator for OpenCRAVAT to improve genomic research by integrating diverse data, including ancestry-specific allele frequencies. You can read more about Moez's experience developing the annotator here: www.opencravat.org/bringing-gen...
#opensource
#opensource
Reposted by Moez Dawood

🚨 Most variant screens measure growth or abundance. What do they miss? That variants impact a spectrum of protein and cellular phenotypes. Variant in situ sequencing (VIS-seq) finds what’s missing: image cells 🔬 first, decode later, revealing multi-scale phenotypes for thousands of variants.👇
1/9
1/9
July 7, 2025 at 2:44 AM
🚨 Most variant screens measure growth or abundance. What do they miss? That variants impact a spectrum of protein and cellular phenotypes. Variant in situ sequencing (VIS-seq) finds what’s missing: image cells 🔬 first, decode later, revealing multi-scale phenotypes for thousands of variants.👇
1/9
1/9
Reposted by Moez Dawood

Excited to share our @varianteffect.bsky.social CVI workstream preprint! Herein, we discuss important considerations for integration of multiplex functional data to generate a single score set and how this is likely to impact variant classification now and in the future
arxiv.org/abs/2503.18810
arxiv.org/abs/2503.18810

Combining multiplexed functional data to improve variant classification
With the surge in the number of variants of uncertain significance (VUS) reported in ClinVar in recent years, there is an imperative to resolve VUS at scale. Multiplexed assays of variant effect (MAVE...
arxiv.org
March 25, 2025 at 3:46 PM
Excited to share our @varianteffect.bsky.social CVI workstream preprint! Herein, we discuss important considerations for integration of multiplex functional data to generate a single score set and how this is likely to impact variant classification now and in the future
arxiv.org/abs/2503.18810
arxiv.org/abs/2503.18810
Reposted by Moez Dawood

Excited to have this out as a preprint!
It was a pleasure to work with @calhoujd.bsky.social and all the coauthors brought together by @varianteffect.bsky.social.
It was a pleasure to work with @calhoujd.bsky.social and all the coauthors brought together by @varianteffect.bsky.social.
Excited to share our @varianteffect.bsky.social CVI workstream preprint! Herein, we discuss important considerations for integration of multiplex functional data to generate a single score set and how this is likely to impact variant classification now and in the future
arxiv.org/abs/2503.18810
arxiv.org/abs/2503.18810
Combining multiplexed functional data to improve variant classification
With the surge in the number of variants of uncertain significance (VUS) reported in ClinVar in recent years, there is an imperative to resolve VUS at scale. Multiplexed assays of variant effect (MAVE...
arxiv.org
March 25, 2025 at 6:32 PM
Excited to have this out as a preprint!
It was a pleasure to work with @calhoujd.bsky.social and all the coauthors brought together by @varianteffect.bsky.social.
It was a pleasure to work with @calhoujd.bsky.social and all the coauthors brought together by @varianteffect.bsky.social.
🚨 Big news at #ACMG2025! 🚨
Today we’re announcing global democratization of deidentified allele count + frequency data with population breakdown from the first ~250k short-read WGS in All of Us designed to plug straight into clinical workflows. It is ~1.1 billion unique variants! 🧬💡
🧵 (1/4)
Today we’re announcing global democratization of deidentified allele count + frequency data with population breakdown from the first ~250k short-read WGS in All of Us designed to plug straight into clinical workflows. It is ~1.1 billion unique variants! 🧬💡
🧵 (1/4)
March 21, 2025 at 2:21 PM
🚨 Big news at #ACMG2025! 🚨
Today we’re announcing global democratization of deidentified allele count + frequency data with population breakdown from the first ~250k short-read WGS in All of Us designed to plug straight into clinical workflows. It is ~1.1 billion unique variants! 🧬💡
🧵 (1/4)
Today we’re announcing global democratization of deidentified allele count + frequency data with population breakdown from the first ~250k short-read WGS in All of Us designed to plug straight into clinical workflows. It is ~1.1 billion unique variants! 🧬💡
🧵 (1/4)
Reposted by Moez Dawood

At #ACMG25? 🧬
Don't miss @moezdawood.bsky.social presenting "GREGoR: Accelerating Genomics for Rare Diseases"
Friday 3/20 at 1:30pm PST
Platform Session 8
Preprint here: pubmed.ncbi.nlm.nih.gov/39764392/
#Genomics #Research #Collaboration @gregor-research.bsky.social
Don't miss @moezdawood.bsky.social presenting "GREGoR: Accelerating Genomics for Rare Diseases"
Friday 3/20 at 1:30pm PST
Platform Session 8
Preprint here: pubmed.ncbi.nlm.nih.gov/39764392/
#Genomics #Research #Collaboration @gregor-research.bsky.social

GREGoR: Accelerating Genomics for Rare Diseases - PubMed
Rare diseases are collectively common, affecting approximately one in twenty individuals worldwide. In recent years, rapid progress has been made in rare disease diagnostics due to advances in DNA seq...
pubmed.ncbi.nlm.nih.gov
March 20, 2025 at 8:21 PM
At #ACMG25? 🧬
Don't miss @moezdawood.bsky.social presenting "GREGoR: Accelerating Genomics for Rare Diseases"
Friday 3/20 at 1:30pm PST
Platform Session 8
Preprint here: pubmed.ncbi.nlm.nih.gov/39764392/
#Genomics #Research #Collaboration @gregor-research.bsky.social
Don't miss @moezdawood.bsky.social presenting "GREGoR: Accelerating Genomics for Rare Diseases"
Friday 3/20 at 1:30pm PST
Platform Session 8
Preprint here: pubmed.ncbi.nlm.nih.gov/39764392/
#Genomics #Research #Collaboration @gregor-research.bsky.social
Reposted by Moez Dawood

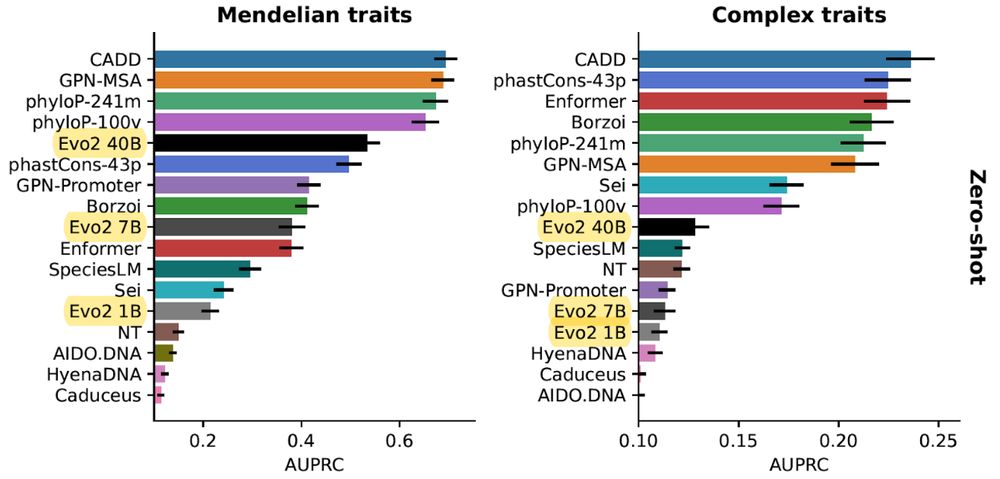
In our updated TraitGym preprint (w/ @gonzalobenegas.bsky.social & Gökcen Eraslan), we evaluate Evo 2 on regulatory variants associated with human traits. We see marked performance gains with scale on Mendelian traits, although still a bit behind alignment-based methods.
doi.org/10.1101/2025...
1/n
doi.org/10.1101/2025...
1/n
March 4, 2025 at 7:54 PM
In our updated TraitGym preprint (w/ @gonzalobenegas.bsky.social & Gökcen Eraslan), we evaluate Evo 2 on regulatory variants associated with human traits. We see marked performance gains with scale on Mendelian traits, although still a bit behind alignment-based methods.
doi.org/10.1101/2025...
1/n
doi.org/10.1101/2025...
1/n
Reposted by Moez Dawood

I'm very happy to share our latest work with Seth Berger and the UCI-GREGoR team. Using long-read sequencing, we can detect de novo variants *without* sequencing both parents. We call our method duoNovo.
preprint: www.medrxiv.org/content/10.1...
R package: github.com/sbergercnmc/...
(1/n)
preprint: www.medrxiv.org/content/10.1...
R package: github.com/sbergercnmc/...
(1/n)
February 27, 2025 at 10:49 PM
I'm very happy to share our latest work with Seth Berger and the UCI-GREGoR team. Using long-read sequencing, we can detect de novo variants *without* sequencing both parents. We call our method duoNovo.
preprint: www.medrxiv.org/content/10.1...
R package: github.com/sbergercnmc/...
(1/n)
preprint: www.medrxiv.org/content/10.1...
R package: github.com/sbergercnmc/...
(1/n)
Reposted by Moez Dawood

#standards, #mechanisms, & #identifiers - oh my!
Just in time for #RareDisease Day, the @gene2phenotype.bsky.social website has a fresh look & new features to improve access to gene-disease models. 🧬💻
👉 Updated website: www.ebi.ac.uk/gene2phenoty...
👉 Why it matters: www.ebi.ac.uk/about/news/u...
Just in time for #RareDisease Day, the @gene2phenotype.bsky.social website has a fresh look & new features to improve access to gene-disease models. 🧬💻
👉 Updated website: www.ebi.ac.uk/gene2phenoty...
👉 Why it matters: www.ebi.ac.uk/about/news/u...
We have launched an updated Gene2Phenotype website with a fresh new look. We now support more detailed disease mechanism information in our expert-curated gene-disease models.
Available at www.ebi.ac.uk/gene2phenotype.
Available at www.ebi.ac.uk/gene2phenotype.
Gene2Phenotype
www.ebi.ac.uk
February 27, 2025 at 8:29 PM
#standards, #mechanisms, & #identifiers - oh my!
Just in time for #RareDisease Day, the @gene2phenotype.bsky.social website has a fresh look & new features to improve access to gene-disease models. 🧬💻
👉 Updated website: www.ebi.ac.uk/gene2phenoty...
👉 Why it matters: www.ebi.ac.uk/about/news/u...
Just in time for #RareDisease Day, the @gene2phenotype.bsky.social website has a fresh look & new features to improve access to gene-disease models. 🧬💻
👉 Updated website: www.ebi.ac.uk/gene2phenoty...
👉 Why it matters: www.ebi.ac.uk/about/news/u...
Reposted by Moez Dawood

Interested in rare variants, the X-chromosome, sex-differences, pharmacogenetics, or transcription factors? You might be interested in our new manuscript where we identified >700 functional rare variants with a difference in effect by sex in GTEx! bit.ly/x_rv_sex #genomics #multiomics 💻🧬

Functional impact of rare variants and sex across the X-chromosome and autosomes
The human X-chromosome contains hundreds of genes and has well-established impacts on sex differences and traits. However, the X-chromosome is often excluded from many genetic analyses, limiting broad...
www.biorxiv.org
January 24, 2025 at 3:58 PM
Interested in rare variants, the X-chromosome, sex-differences, pharmacogenetics, or transcription factors? You might be interested in our new manuscript where we identified >700 functional rare variants with a difference in effect by sex in GTEx! bit.ly/x_rv_sex #genomics #multiomics 💻🧬
Reposted by Moez Dawood

A map of the rubisco biochemical landscape.
Rubisco is the main CO2-fixing enzyme of the biosphere yet its kinetics are slow. This work shows that non-trivial biochemical changes are readily accessible pointing the way to new engineering ways
🧪 @nature.com
www.nature.com/articles/s41...
Rubisco is the main CO2-fixing enzyme of the biosphere yet its kinetics are slow. This work shows that non-trivial biochemical changes are readily accessible pointing the way to new engineering ways
🧪 @nature.com
www.nature.com/articles/s41...

A map of the rubisco biochemical landscape - Nature
A massively parallel assay developed to map the essential photosynthetic enzyme rubisco showed that non-trivial biochemical changes and improvements in CO2 affinity are possible, signposting further e...
www.nature.com
January 24, 2025 at 4:02 PM
A map of the rubisco biochemical landscape.
Rubisco is the main CO2-fixing enzyme of the biosphere yet its kinetics are slow. This work shows that non-trivial biochemical changes are readily accessible pointing the way to new engineering ways
🧪 @nature.com
www.nature.com/articles/s41...
Rubisco is the main CO2-fixing enzyme of the biosphere yet its kinetics are slow. This work shows that non-trivial biochemical changes are readily accessible pointing the way to new engineering ways
🧪 @nature.com
www.nature.com/articles/s41...
Reposted by Moez Dawood
Proud to introduce curated loci prime editing (cliPE)! It is a generalizable and accessible platform to characterize many genetic variants in a gene of interest 1/11
arxiv.org/abs/2501.04822
arxiv.org/abs/2501.04822

Curated loci prime editing (cliPE) for accessible multiplexed assays of variant effect (MAVEs)
Multiplexed assays of variant effect (MAVEs) perform simultaneous characterization of many variants. Prime editing has been recently adopted for introducing many variants in their native genomic conte...
arxiv.org
January 10, 2025 at 6:49 PM
Proud to introduce curated loci prime editing (cliPE)! It is a generalizable and accessible platform to characterize many genetic variants in a gene of interest 1/11
arxiv.org/abs/2501.04822
arxiv.org/abs/2501.04822
Reposted by Moez Dawood

What if one variant can cause splicing outliers transcriptome-wide? In our preprint, we show how examining transcriptome-wide patterns of splicing outliers can both diagnose individuals with rare spliceopathies and uncover novel disease-gene relationships! (www.medrxiv.org/content/10.1...)

Transcriptome-wide outlier approach identifies individuals with minor spliceopathies
RNA-sequencing has improved the diagnostic yield of individuals with rare diseases. Current analyses predominantly focus on identifying outliers in single genes that can be attributed to cis-acting va...
www.medrxiv.org
January 7, 2025 at 9:15 PM
What if one variant can cause splicing outliers transcriptome-wide? In our preprint, we show how examining transcriptome-wide patterns of splicing outliers can both diagnose individuals with rare spliceopathies and uncover novel disease-gene relationships! (www.medrxiv.org/content/10.1...)
Reposted by Moez Dawood

GREGoR Consortium preprint online! With the R02 release, there is a huge amount of genomics, multi-omics and phenotype data from the hardest-to-solve rare disease cases.
🚨 Excited to announce the Marker paper for the GREGoR Consortium! arxiv.org/abs/2412.14338
Accelerating #RareDisease diagnostics with cutting-edge #Genomics and global data sharing of omics and deep phenotyping from ~7500 individuals on NHGRI AnVIL and much more to come! 🧬
Accelerating #RareDisease diagnostics with cutting-edge #Genomics and global data sharing of omics and deep phenotyping from ~7500 individuals on NHGRI AnVIL and much more to come! 🧬

GREGoR: Accelerating Genomics for Rare Diseases
Rare diseases are collectively common, affecting approximately one in twenty individuals worldwide. In recent years, rapid progress has been made in rare disease diagnostics due to advances in DNA seq...
arxiv.org
December 20, 2024 at 2:59 AM
GREGoR Consortium preprint online! With the R02 release, there is a huge amount of genomics, multi-omics and phenotype data from the hardest-to-solve rare disease cases.
🚨 Excited to announce the Marker paper for the GREGoR Consortium! arxiv.org/abs/2412.14338
Accelerating #RareDisease diagnostics with cutting-edge #Genomics and global data sharing of omics and deep phenotyping from ~7500 individuals on NHGRI AnVIL and much more to come! 🧬
Accelerating #RareDisease diagnostics with cutting-edge #Genomics and global data sharing of omics and deep phenotyping from ~7500 individuals on NHGRI AnVIL and much more to come! 🧬
GREGoR: Accelerating Genomics for Rare Diseases
Rare diseases are collectively common, affecting approximately one in twenty individuals worldwide. In recent years, rapid progress has been made in rare disease diagnostics due to advances in DNA seq...
arxiv.org
December 20, 2024 at 1:49 AM
🚨 Excited to announce the Marker paper for the GREGoR Consortium! arxiv.org/abs/2412.14338
Accelerating #RareDisease diagnostics with cutting-edge #Genomics and global data sharing of omics and deep phenotyping from ~7500 individuals on NHGRI AnVIL and much more to come! 🧬
Accelerating #RareDisease diagnostics with cutting-edge #Genomics and global data sharing of omics and deep phenotyping from ~7500 individuals on NHGRI AnVIL and much more to come! 🧬
Reposted by Moez Dawood

Genomic duplications involving MECP2 are surprisingly complex and affect almost half of patients. Importantly, complexity contributes to gene expression and clinical variability. @pnrigenetics.bsky.social @gregor-research.bsky.social genomemedicine.biomedcentral.com/articles/10....

Structural variant allelic heterogeneity in MECP2 duplication syndrome provides insight into clinical severity and variability of disease expression - Genome Medicine
Background MECP2 Duplication Syndrome, also known as X-linked intellectual developmental disorder Lubs type (MRXSL; MIM: 300260), is a neurodevelopmental disorder caused by copy number gains spanning ...
genomemedicine.biomedcentral.com
December 18, 2024 at 9:43 PM
Genomic duplications involving MECP2 are surprisingly complex and affect almost half of patients. Importantly, complexity contributes to gene expression and clinical variability. @pnrigenetics.bsky.social @gregor-research.bsky.social genomemedicine.biomedcentral.com/articles/10....
Paper came out this past week! Using MAVEs to reduce variant classification disparities in underrepresented populations and demonstrating AI bias in computational predictors rdcu.be/d2kCn
Previous tweetorial: x.com/MoezDawood/s...
Previous post by @ee-reh-neh.bsky.social: bsky.app/profile/ee-r...
Previous tweetorial: x.com/MoezDawood/s...
Previous post by @ee-reh-neh.bsky.social: bsky.app/profile/ee-r...
December 8, 2024 at 3:19 PM
Paper came out this past week! Using MAVEs to reduce variant classification disparities in underrepresented populations and demonstrating AI bias in computational predictors rdcu.be/d2kCn
Previous tweetorial: x.com/MoezDawood/s...
Previous post by @ee-reh-neh.bsky.social: bsky.app/profile/ee-r...
Previous tweetorial: x.com/MoezDawood/s...
Previous post by @ee-reh-neh.bsky.social: bsky.app/profile/ee-r...
Reposted by Moez Dawood

New work! Wherein we (as in, the AVE ODIC working group) looked at clinical variant classification across genetic ancestry groups in gnomAD and AoU and what we found... is exactly what you might expect, after decades of Eurocentric research. But we also show there's a better way forward! 🧬🖥️

Defining and Reducing Variant Classification Disparities https://www.medrxiv.org/content/10.1101/2024.04.11.24305690v1

Defining and Reducing Variant Classification Disparities https://www.medrxiv.org/content/10.1101/2024.04.11.24305690v1
Background: Multiplexed Assays of Variant Effects (MAVEs) can test all possible single variants in a
www.medrxiv.org
April 18, 2024 at 11:07 PM
New work! Wherein we (as in, the AVE ODIC working group) looked at clinical variant classification across genetic ancestry groups in gnomAD and AoU and what we found... is exactly what you might expect, after decades of Eurocentric research. But we also show there's a better way forward! 🧬🖥️