



Adil Kabylda
@kabylda.bsky.social
PhD Researcher in Tkatchenko Group @uni_lu / #LINO22 / BSc and MSc in Chemistry @MSU_1755 @QPD_Lab / Pavlodar KZ 🇰🇿
kabylda.github.io
kabylda.github.io
Reposted by Adil Kabylda

Another cover from our latest issue of #JACS: "Molecular Simulations with a Pretrained Neural Network and Universal Pairwise Force Fields"
Explore the research behind the art 🔗: buff.ly/J3R4JpL
#ChemSky
Explore the research behind the art 🔗: buff.ly/J3R4JpL
#ChemSky

September 19, 2025 at 12:01 PM
Another cover from our latest issue of #JACS: "Molecular Simulations with a Pretrained Neural Network and Universal Pairwise Force Fields"
Explore the research behind the art 🔗: buff.ly/J3R4JpL
#ChemSky
Explore the research behind the art 🔗: buff.ly/J3R4JpL
#ChemSky
Our recent work on SO3LR, a general-purpose machine learned force field for molecular simulations, has been published in @jacs.acspublications.org! 🌞 doi.org/10.1021/jacs...

September 7, 2025 at 4:24 PM
Our recent work on SO3LR, a general-purpose machine learned force field for molecular simulations, has been published in @jacs.acspublications.org! 🌞 doi.org/10.1021/jacs...
Reposted by Adil Kabylda

Martini 3 - IDP is out! Improved parameters for disordered proteins, great work from Liguo Wang: www.nature.com/articles/s41...
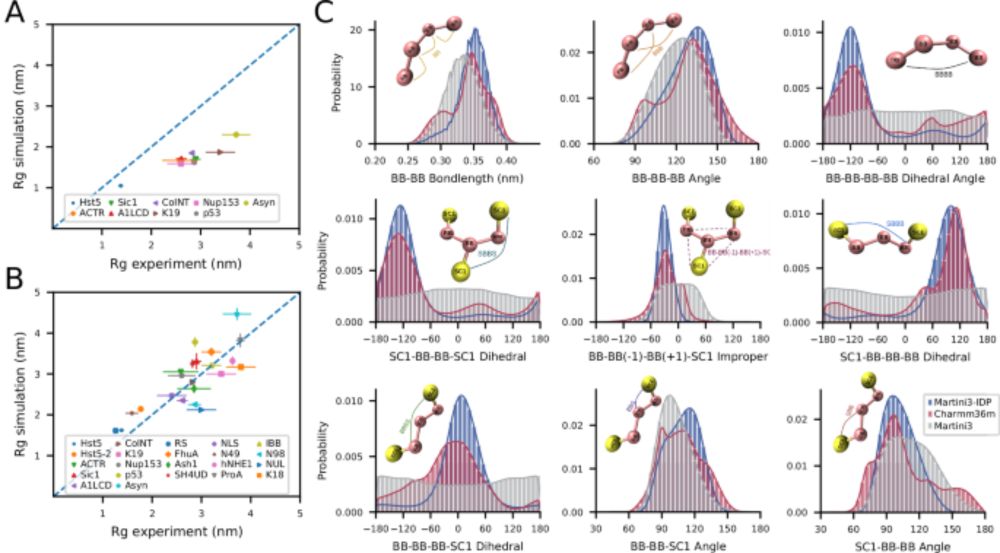
Martini3-IDP: improved Martini 3 force field for disordered proteins - Nature Communications
Here, the authors introduce Martini3-IDP, a refined model for disordered proteins that addresses prior over-compact structures. Validated across diverse systems, it captures IDP interactions and biomo...
www.nature.com
March 24, 2025 at 8:09 PM
Martini 3 - IDP is out! Improved parameters for disordered proteins, great work from Liguo Wang: www.nature.com/articles/s41...
Reposted by Adil Kabylda

Simulating full quantum mechanical ground- and excited state surfaces with deep quantum Monte Carlo by Zeno Schätzle, Bernat Szabo and Alice Cuzzocrea.
arxiv.org/abs/2503.19847
🧵⬇️
arxiv.org/abs/2503.19847
🧵⬇️

March 26, 2025 at 10:45 AM
Simulating full quantum mechanical ground- and excited state surfaces with deep quantum Monte Carlo by Zeno Schätzle, Bernat Szabo and Alice Cuzzocrea.
arxiv.org/abs/2503.19847
🧵⬇️
arxiv.org/abs/2503.19847
🧵⬇️
Reposted by Adil Kabylda

This is a remarkable paper! A gigantic dataset of highly precise, highly accurate first-principles data. This builds on years of work on @fhi-aims.bsky.social - enabling dispersion-corrected hybrid DFT that covers a huge swath of chemical space. Congrats to the authors!
doi.org/10.1038/s415...
doi.org/10.1038/s415...
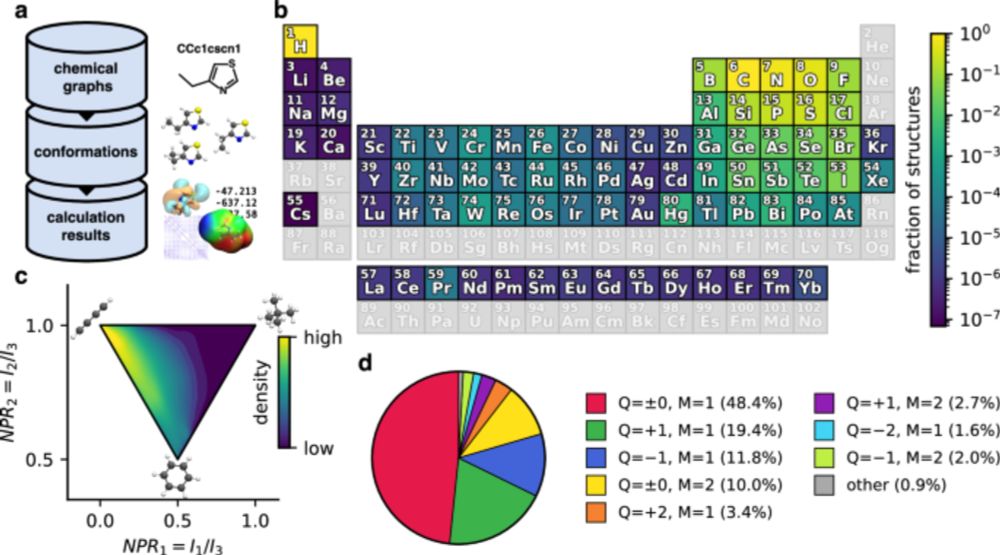
The QCML dataset, Quantum chemistry reference data from 33.5M DFT and 14.7B semi-empirical calculations - Scientific Data
Scientific Data - The QCML dataset, Quantum chemistry reference data from 33.5M DFT and 14.7B semi-empirical calculations
urldefense.com
March 12, 2025 at 8:03 PM
This is a remarkable paper! A gigantic dataset of highly precise, highly accurate first-principles data. This builds on years of work on @fhi-aims.bsky.social - enabling dispersion-corrected hybrid DFT that covers a huge swath of chemical space. Congrats to the authors!
doi.org/10.1038/s415...
doi.org/10.1038/s415...
Reposted by Adil Kabylda

I rarely have been more excited about one of our papers. Come join the PET-MADness, to get better #ML potentials with less data 📈!

📢 PET-MAD has just landed! 📢 What if I told you that you can match & improve the accuracy of other "universal" #machinelearning potentials training on fewer than 100k atomic structures? And be *faster* with an unconstrained architecture that is conservative with tiny symmetry breaking? Sounds like 🧑🚀
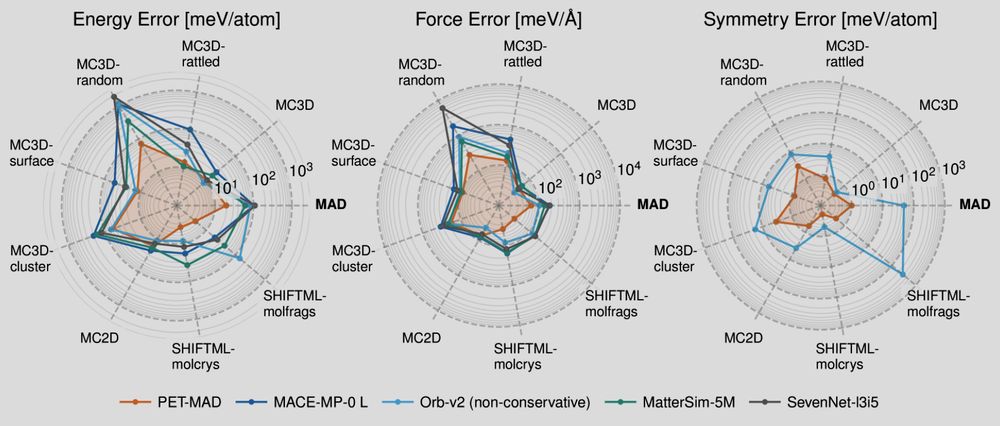
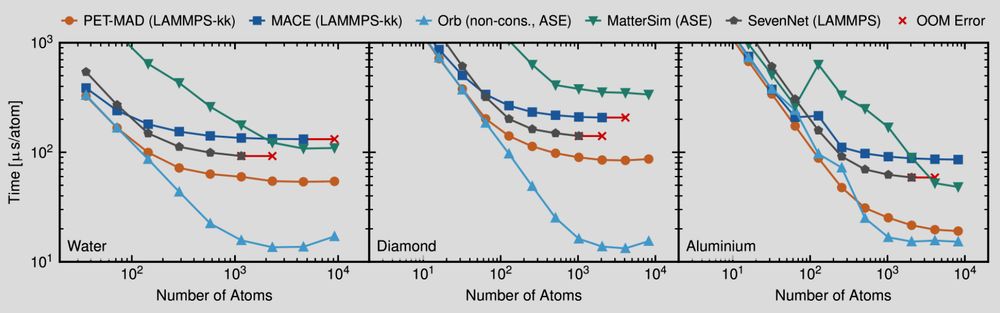
March 19, 2025 at 8:41 AM
I rarely have been more excited about one of our papers. Come join the PET-MADness, to get better #ML potentials with less data 📈!
Reposted by Adil Kabylda

#compchem Good read: CHARMM-GUI EnzyDocker for Protein–Ligand Docking of Multiple Reactive States along a Reaction Coordinate in Enzymes #compchemsky #biosky pubs.acs.org/doi/10.1021/...

CHARMM-GUI EnzyDocker for Protein–Ligand Docking of Multiple Reactive States along a Reaction Coordinate in Enzymes
Enzymes play crucial roles in all biological systems by catalyzing a myriad of chemical reactions. These reactions range from simple one-step processes to intricate multistep cascades. Predicting mech...
pubs.acs.org
February 21, 2025 at 6:48 AM
#compchem Good read: CHARMM-GUI EnzyDocker for Protein–Ligand Docking of Multiple Reactive States along a Reaction Coordinate in Enzymes #compchemsky #biosky pubs.acs.org/doi/10.1021/...
Reposted by Adil Kabylda

”Models that deviate too far from known physical principles produce unstable MD trajectories, even when they have very high energy and force prediction accuracy.”
doi.org/10.1021/acs....
doi.org/10.1021/acs....

Analyzing Atomic Interactions in Molecules as Learned by Neural Networks
While machine learning (ML) models have been able to achieve unprecedented accuracies across various prediction tasks in quantum chemistry, it is now apparent that accuracy on a test set alone is not a guarantee for robust chemical modeling such as stable molecular dynamics (MD). To go beyond accuracy, we use explainable artificial intelligence (XAI) techniques to develop a general analysis framework for atomic interactions and apply it to the SchNet and PaiNN neural network models. We compare these interactions with a set of fundamental chemical principles to understand how well the models have learned the underlying physicochemical concepts from the data. We focus on the strength of the interactions for different atomic species, how predictions for intensive and extensive quantum molecular properties are made, and analyze the decay and many-body nature of the interactions with interatomic distance. Models that deviate too far from known physical principles produce unstable MD trajectories, even when they have very high energy and force prediction accuracy. We also suggest further improvements to the ML architectures to better account for the polynomial decay of atomic interactions.
doi.org
January 12, 2025 at 10:18 PM
”Models that deviate too far from known physical principles produce unstable MD trajectories, even when they have very high energy and force prediction accuracy.”
doi.org/10.1021/acs....
doi.org/10.1021/acs....
Reposted by Adil Kabylda

A fascinating paper elucidating ion conduction in a potassium channel experimentally
www.sciencedirect.com/science/arti...
www.sciencedirect.com/science/arti...

Direct visualization of electric-field-stimulated ion conduction in a potassium channel
Understanding protein function would be facilitated by direct, real-time observation of chemical kinetics in the atomic structure. The selectivity fil…
www.sciencedirect.com
January 10, 2025 at 11:24 AM
A fascinating paper elucidating ion conduction in a potassium channel experimentally
www.sciencedirect.com/science/arti...
www.sciencedirect.com/science/arti...
Reposted by Adil Kabylda

We are seeking an outstanding #PhD candidate to join our team (www.duartegroupchem.org)! If you know potential candidates interested in combining #ML and #compchem reaction modelling for catalyst design, please share this opportunity with them!
🗓 Deadline: 31/01/25
📥 Application: shorturl.at/nHyAo
🗓 Deadline: 31/01/25
📥 Application: shorturl.at/nHyAo

January 5, 2025 at 9:33 AM
We are seeking an outstanding #PhD candidate to join our team (www.duartegroupchem.org)! If you know potential candidates interested in combining #ML and #compchem reaction modelling for catalyst design, please share this opportunity with them!
🗓 Deadline: 31/01/25
📥 Application: shorturl.at/nHyAo
🗓 Deadline: 31/01/25
📥 Application: shorturl.at/nHyAo
Reposted by Adil Kabylda

Excited to share our latest work on Euclidean fast attention, which enables learning global atomic representations at linear cost! 🔥
The representations describe the distance and orientation between atoms, crucial for modeling molecular systems
tinyurl.com/47xud8nr
#MachineLearning #AI4Science
The representations describe the distance and orientation between atoms, crucial for modeling molecular systems
tinyurl.com/47xud8nr
#MachineLearning #AI4Science
Euclidean Fast Attention: Machine Learning Global Atomic Representations at Linear Cost
Long-range correlations are essential across numerous machine learning tasks, especially for data embedded in Euclidean space, where the relative positions and orientations of distant components are o...
tinyurl.com
January 3, 2025 at 8:50 PM
Excited to share our latest work on Euclidean fast attention, which enables learning global atomic representations at linear cost! 🔥
The representations describe the distance and orientation between atoms, crucial for modeling molecular systems
tinyurl.com/47xud8nr
#MachineLearning #AI4Science
The representations describe the distance and orientation between atoms, crucial for modeling molecular systems
tinyurl.com/47xud8nr
#MachineLearning #AI4Science
Reposted by Adil Kabylda

SPINACH ON THE CEILING: A Theoretical Chemist's Return to Biology by Martin Karplus. #NMRchat #chemsky
www.annualreviews.org/content/jour...
December 31, 2024 at 3:37 PM
SPINACH ON THE CEILING: A Theoretical Chemist's Return to Biology by Martin Karplus. #NMRchat #chemsky
www.annualreviews.org/content/jour...