



Jean P. Elbers
@jpelbers.bsky.social
Lead Bioinformatician
Nanopore Sequencing Lab
Institute of Medical Genetics
Medical University of Vienna
Nanopore Sequencing Lab
Institute of Medical Genetics
Medical University of Vienna

Reposted by Jean P. Elbers



October 30, 2025 at 7:27 PM
Reposted by Jean P. Elbers

The enduring advantages of the SLOW5 file format for raw nanopore sequencing data. #Nanopore #Sequencing #SLOW5 #DataFile #Genomics #Bioinformatics @gigascience.bsky.social @nanoporetech.com
academic.oup.com/gigascience/...
academic.oup.com/gigascience/...

October 25, 2025 at 7:05 PM
The enduring advantages of the SLOW5 file format for raw nanopore sequencing data. #Nanopore #Sequencing #SLOW5 #DataFile #Genomics #Bioinformatics @gigascience.bsky.social @nanoporetech.com
academic.oup.com/gigascience/...
academic.oup.com/gigascience/...
Reposted by Jean P. Elbers

This makes surprising amount of sense.

October 25, 2025 at 2:22 AM
This makes surprising amount of sense.
Reposted by Jean P. Elbers

Ever wanted to get the query sequence from a bam using bed coordinates?
How about the matching coordinates of an assembly mapped to a reference but repeats and SVs are confusing liftover?
Well I did while trying to benchmark STR stuff. Nice side quest.
github.com/Psy-Fer/bedp...
How about the matching coordinates of an assembly mapped to a reference but repeats and SVs are confusing liftover?
Well I did while trying to benchmark STR stuff. Nice side quest.
github.com/Psy-Fer/bedp...

GitHub - Psy-Fer/bedpull: bedpull - Pull the query sequence from bam or fasta references using a bed file
bedpull - Pull the query sequence from bam or fasta references using a bed file - Psy-Fer/bedpull
github.com
October 23, 2025 at 3:59 AM
Ever wanted to get the query sequence from a bam using bed coordinates?
How about the matching coordinates of an assembly mapped to a reference but repeats and SVs are confusing liftover?
Well I did while trying to benchmark STR stuff. Nice side quest.
github.com/Psy-Fer/bedp...
How about the matching coordinates of an assembly mapped to a reference but repeats and SVs are confusing liftover?
Well I did while trying to benchmark STR stuff. Nice side quest.
github.com/Psy-Fer/bedp...
Reposted by Jean P. Elbers

Using renv::restore() and it seemingly takes a hundred years to download packages?
Try
options(renv.config.pak.enabled = TRUE)
which will use pak to download and install packages *in parallel*. So much faster.
Try
options(renv.config.pak.enabled = TRUE)
which will use pak to download and install packages *in parallel*. So much faster.
September 25, 2025 at 8:01 AM
Using renv::restore() and it seemingly takes a hundred years to download packages?
Try
options(renv.config.pak.enabled = TRUE)
which will use pak to download and install packages *in parallel*. So much faster.
Try
options(renv.config.pak.enabled = TRUE)
which will use pak to download and install packages *in parallel*. So much faster.
Reposted by Jean P. Elbers

Prototyping #rshiny apps to native #electron desktop apps:
shinyelectron::export() → #rshinylive conversion → .dmg → Native Mac app
Zero #rstats dependencies for end users! Early days but promising 👀
shinyelectron::export() → #rshinylive conversion → .dmg → Native Mac app
Zero #rstats dependencies for end users! Early days but promising 👀
September 4, 2025 at 7:35 AM
Prototyping #rshiny apps to native #electron desktop apps:
shinyelectron::export() → #rshinylive conversion → .dmg → Native Mac app
Zero #rstats dependencies for end users! Early days but promising 👀
shinyelectron::export() → #rshinylive conversion → .dmg → Native Mac app
Zero #rstats dependencies for end users! Early days but promising 👀
Reposted by Jean P. Elbers

Thank you Tommy!
The link you gave is good but points to a 2yr old rendering. This one uses recent versions of R and all packages and has all the latest typo fixes:
www.huber.embl.de/msmb/
The link you gave is good but points to a 2yr old rendering. This one uses recent versions of R and all packages and has all the latest typo fixes:
www.huber.embl.de/msmb/

Modern Statistics for Modern Biology
If you are a biologist and want to get the best out of the powerful methods of modern computational statistics, this is your book.
www.huber.embl.de
August 29, 2025 at 1:48 PM
Thank you Tommy!
The link you gave is good but points to a 2yr old rendering. This one uses recent versions of R and all packages and has all the latest typo fixes:
www.huber.embl.de/msmb/
The link you gave is good but points to a 2yr old rendering. This one uses recent versions of R and all packages and has all the latest typo fixes:
www.huber.embl.de/msmb/
Reposted by Jean P. Elbers

☺️

Dr Hasindu Gamaarachchi @hasindu2008.bsky.social @unsw.edu.au @garvaninstitute.bsky.social is a finalist in the Macquarie University Eureka Prize for Outstanding Early Career Researcher.
Learn more: youtu.be/FL8ZKUmtYbM
#EurekaPrizes
Learn more: youtu.be/FL8ZKUmtYbM
#EurekaPrizes

August 3, 2025 at 9:04 AM
☺️
Reposted by Jean P. Elbers

Yes, it has worked really well. I should mention that I'm porting to the Godot game engine. Zig and Claude work great together, but Godot brings some advantages that outweigh how well they work together. Godot brings WebGL support, platform pipeline, visual UI editor, and some debugging tools.
August 1, 2025 at 5:59 AM
Yes, it has worked really well. I should mention that I'm porting to the Godot game engine. Zig and Claude work great together, but Godot brings some advantages that outweigh how well they work together. Godot brings WebGL support, platform pipeline, visual UI editor, and some debugging tools.
Reposted by Jean P. Elbers
If you are at #ISMB2025:
@bosc.bsky.social track around 2:30pm ish after
@sunethsa.bsky.social's talk, Bonson Wong will present on
nanopore basecalling on AMD GPUs using slorado
github.com/BonsonW/slor...
@bosc.bsky.social track around 2:30pm ish after
@sunethsa.bsky.social's talk, Bonson Wong will present on
nanopore basecalling on AMD GPUs using slorado
github.com/BonsonW/slor...
GitHub - BonsonW/slorado: A simplified version of Dorado built on top of S/BLOW5 format.
A simplified version of Dorado built on top of S/BLOW5 format. - BonsonW/slorado
github.com
July 21, 2025 at 9:03 AM
If you are at #ISMB2025:
@bosc.bsky.social track around 2:30pm ish after
@sunethsa.bsky.social's talk, Bonson Wong will present on
nanopore basecalling on AMD GPUs using slorado
github.com/BonsonW/slor...
@bosc.bsky.social track around 2:30pm ish after
@sunethsa.bsky.social's talk, Bonson Wong will present on
nanopore basecalling on AMD GPUs using slorado
github.com/BonsonW/slor...
Reposted by Jean P. Elbers

There is also a branch that takes nanopore reads as input, which works reasonably well. We are putting some final code touches on it, but maybe helpful - github.com/wwood/single...

Nanopore by thepatientwait · Pull Request #208 · wwood/singlem
Working Nanopore build.
Important changes:
DIAMOND prefilter
Uses --range-culling + related args for DIAMOND.
These results are now streamed to help memory.
Sequences are indexed using gene names ...
github.com
July 18, 2025 at 1:07 AM
There is also a branch that takes nanopore reads as input, which works reasonably well. We are putting some final code touches on it, but maybe helpful - github.com/wwood/single...
Reposted by Jean P. Elbers

OrthoFinder just dropped a major update
It’s faster, more accurate, and ready for thousands of genomes
Let’s break it down (1/10)
github.com/OrthoFinder/...
www.biorxiv.org/content/10.1...
It’s faster, more accurate, and ready for thousands of genomes
Let’s break it down (1/10)
github.com/OrthoFinder/...
www.biorxiv.org/content/10.1...
July 16, 2025 at 5:51 PM
OrthoFinder just dropped a major update
It’s faster, more accurate, and ready for thousands of genomes
Let’s break it down (1/10)
github.com/OrthoFinder/...
www.biorxiv.org/content/10.1...
It’s faster, more accurate, and ready for thousands of genomes
Let’s break it down (1/10)
github.com/OrthoFinder/...
www.biorxiv.org/content/10.1...
Reposted by Jean P. Elbers

Comprehensive taxonomic identification of microbial species in metagenomic data using SingleM and Sandpiper www.nature.com/articles/s41... #jcampubs
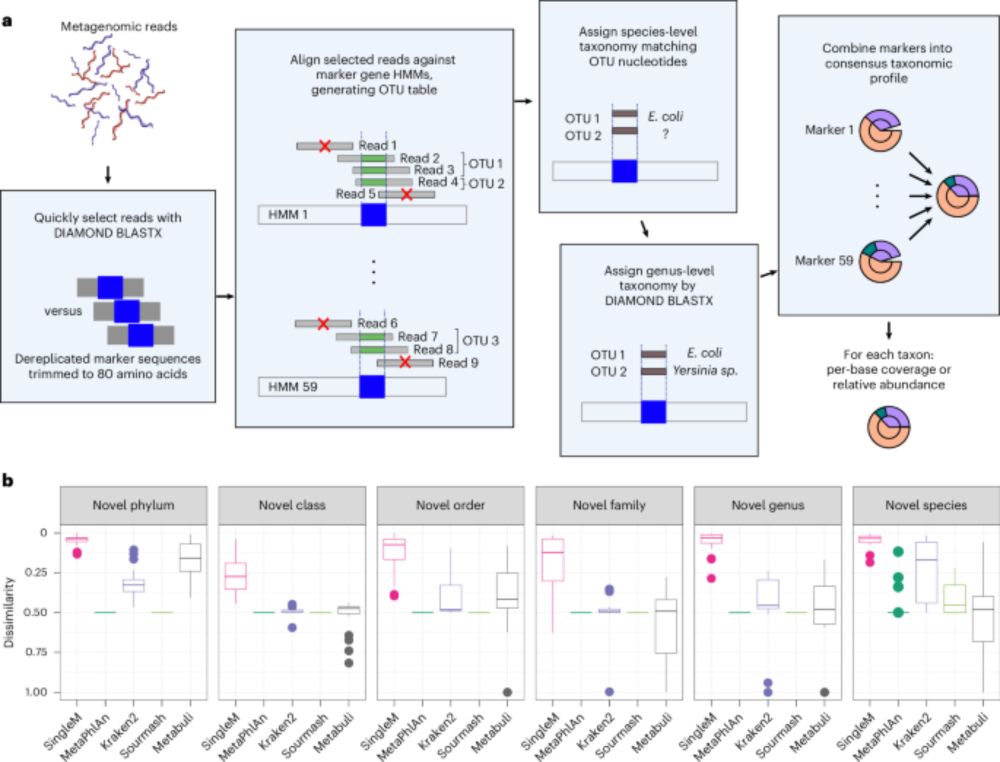
Comprehensive taxonomic identification of microbial species in metagenomic data using SingleM and Sandpiper - Nature Biotechnology
Novel microbial species in metagenomes are identified using conserved regions within universal marker genes.
www.nature.com
July 16, 2025 at 5:21 PM
Comprehensive taxonomic identification of microbial species in metagenomic data using SingleM and Sandpiper www.nature.com/articles/s41... #jcampubs
Reposted by Jean P. Elbers

orbital is such a cool idea — convert your tidymodels or (now) scikit learn model predictions to SQL so you can run them in the database

Announcing Orbital for Python! For Scikit-learn users, this tool transforms your ML pipelines into SQL queries, letting predictions run directly in your database without a #Python environment.
Learn more: posit.co/blog/introdu...
Learn more: posit.co/blog/introdu...

July 14, 2025 at 2:41 PM
orbital is such a cool idea — convert your tidymodels or (now) scikit learn model predictions to SQL so you can run them in the database
Reposted by Jean P. Elbers
We've been developing a small standalone tool for viewing & calculating frequency from modification tags in BAM files. This call is for brave users to test.
github.com/warp9seq/min...
written by
@sunethsa.bsky.social
in C, based on mod tag parsing we did for realfreq doi.org/10.1093/bioi...
github.com/warp9seq/min...
written by
@sunethsa.bsky.social
in C, based on mod tag parsing we did for realfreq doi.org/10.1093/bioi...
GitHub - warp9seq/minimod: A bioinformatics tool for viewing and calculating base modification frequencies from BAM files
A bioinformatics tool for viewing and calculating base modification frequencies from BAM files - warp9seq/minimod
github.com
July 16, 2025 at 6:16 AM
We've been developing a small standalone tool for viewing & calculating frequency from modification tags in BAM files. This call is for brave users to test.
github.com/warp9seq/min...
written by
@sunethsa.bsky.social
in C, based on mod tag parsing we did for realfreq doi.org/10.1093/bioi...
github.com/warp9seq/min...
written by
@sunethsa.bsky.social
in C, based on mod tag parsing we did for realfreq doi.org/10.1093/bioi...
Reposted by Jean P. Elbers
Genome Evaluation Pipeline (GEP): A fully-automated quality control tool for parallel evaluation of genome assemblies. #GenomeAssembly #QualityControl #Snakemake #Genomics #Bioinformatics @bioinfoadv.bsky.social 🧬 🖥️
academic.oup.com/bioinformati...
academic.oup.com/bioinformati...

July 10, 2025 at 2:31 PM
Genome Evaluation Pipeline (GEP): A fully-automated quality control tool for parallel evaluation of genome assemblies. #GenomeAssembly #QualityControl #Snakemake #Genomics #Bioinformatics @bioinfoadv.bsky.social 🧬 🖥️
academic.oup.com/bioinformati...
academic.oup.com/bioinformati...
Reposted by Jean P. Elbers
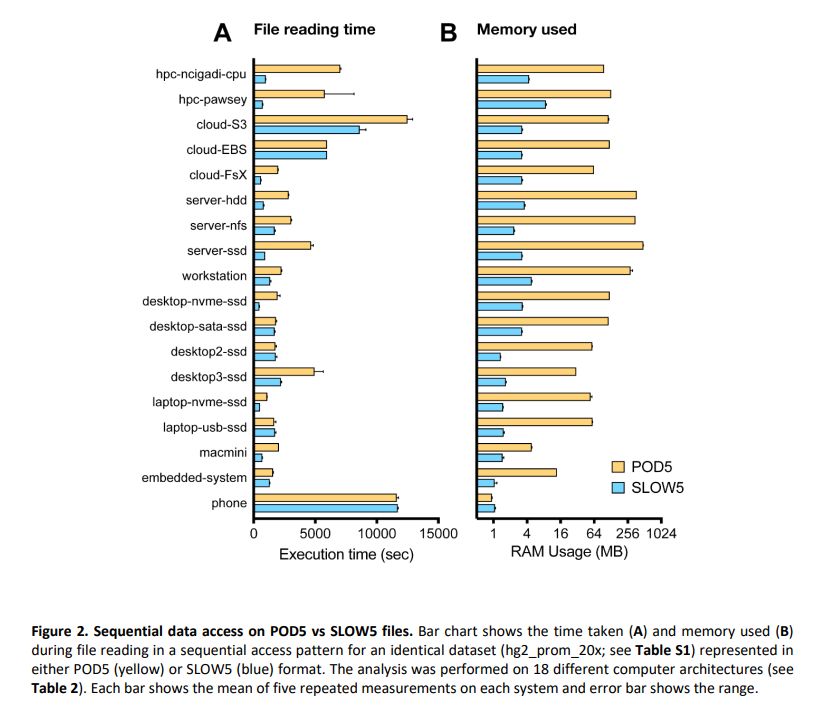
For many of those who were asking on BLOW5 vs POD5 for nanopore signal data, here is a finally detailed benchmark we did:
biorxiv.org/content/10.1...
Summary: performance of BLOW5 is >= POD5 (from ~= to 100X, see below), with benefit of having ~3 dependencies instead of >50.
biorxiv.org/content/10.1...
Summary: performance of BLOW5 is >= POD5 (from ~= to 100X, see below), with benefit of having ~3 dependencies instead of >50.
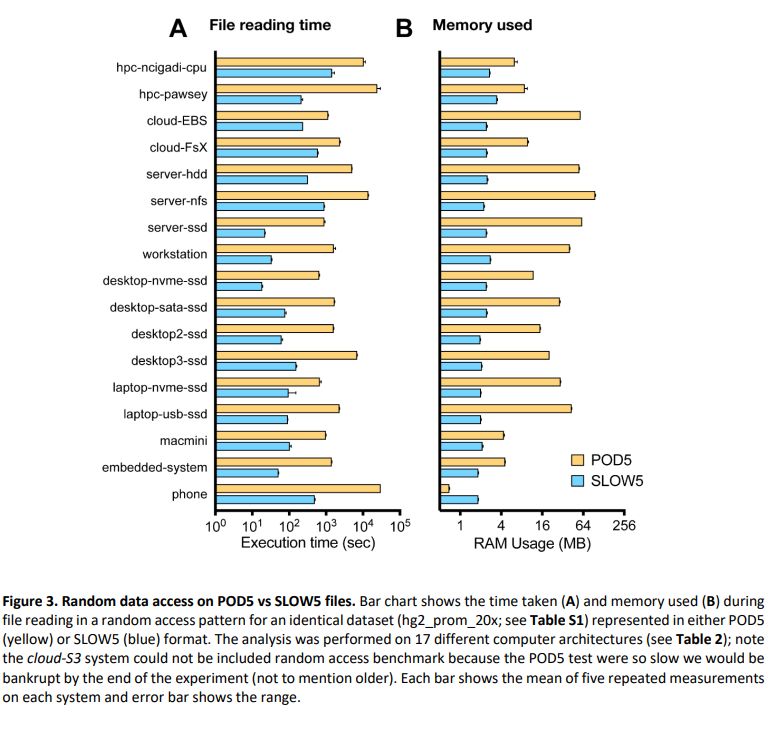
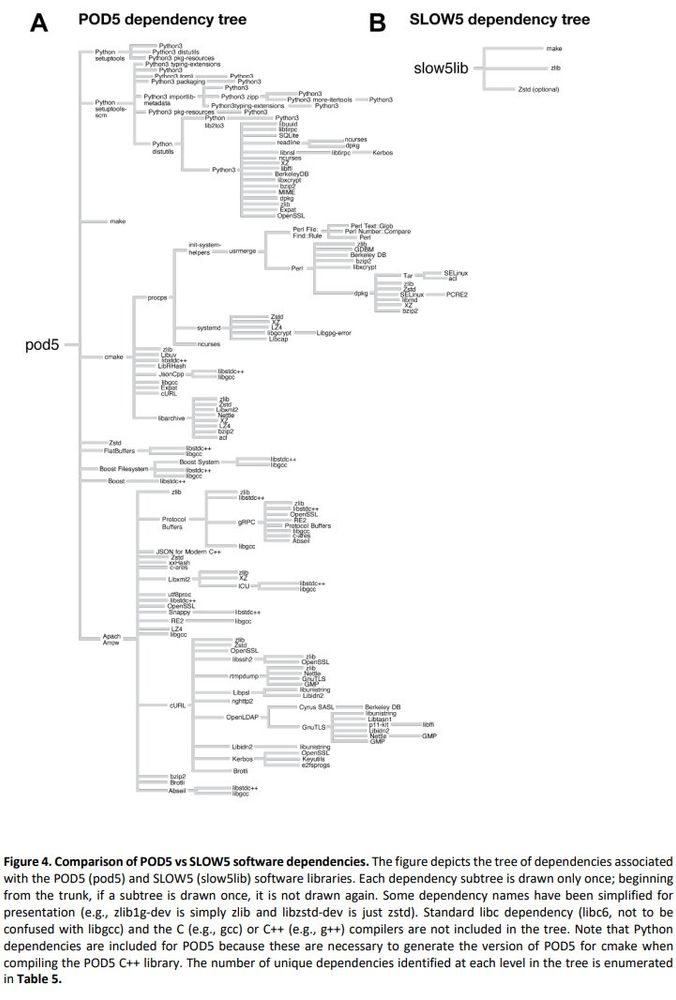
July 5, 2025 at 4:26 AM
For many of those who were asking on BLOW5 vs POD5 for nanopore signal data, here is a finally detailed benchmark we did:
biorxiv.org/content/10.1...
Summary: performance of BLOW5 is >= POD5 (from ~= to 100X, see below), with benefit of having ~3 dependencies instead of >50.
biorxiv.org/content/10.1...
Summary: performance of BLOW5 is >= POD5 (from ~= to 100X, see below), with benefit of having ~3 dependencies instead of >50.
Reposted by Jean P. Elbers

Parallel Self-Hosted Code Generation in the Zig Compiler
https://ziglang.org/devlog/2025/#2025-06-14
https://ziglang.org/devlog/2025/#2025-06-14
Devlog
⚡
Zig Programming Language
https://ziglang.org/devlog/2025/#2025-06-14
ziglang.org
June 14, 2025 at 7:17 PM
Parallel Self-Hosted Code Generation in the Zig Compiler
https://ziglang.org/devlog/2025/#2025-06-14
https://ziglang.org/devlog/2025/#2025-06-14
Reposted by Jean P. Elbers

Announcing myloasm, a new long-read (ONT R10/PacBio) metagenome assembler that I've been working on during my postdoc in the Heng Li lab (@lh3lh3.bsky.social).
myloasm-docs.github.io
myloasm-docs.github.io
myloasm - metagenomic assembly with (noisy) long reads
myloasm-docs.github.io
May 28, 2025 at 5:54 PM
Announcing myloasm, a new long-read (ONT R10/PacBio) metagenome assembler that I've been working on during my postdoc in the Heng Li lab (@lh3lh3.bsky.social).
myloasm-docs.github.io
myloasm-docs.github.io
Reposted by Jean P. Elbers
Preprint on hifiasm Nanopore-only assembly. Led by Haoyu Cheng: www.biorxiv.org/content/10.1...

Efficient near telomere-to-telomere assembly of Nanopore Simplex reads
Telomere-to-telomere (T2T) assembly is the ultimate goal for de novo genome assembly. Existing algorithms capable of near T2T assembly all require Oxford Nanopore Technologies (ONT) ultra-long reads w...
www.biorxiv.org
April 18, 2025 at 9:54 PM
Preprint on hifiasm Nanopore-only assembly. Led by Haoyu Cheng: www.biorxiv.org/content/10.1...
Reposted by Jean P. Elbers

scnanoseq nf-co.re/scrnaseq: an nf-core [Nextflow] pipeline for Oxford Nanopore single-cell RNA-sequencing www.biorxiv.org/content/10.1... 🧬🖥️🧪 github.com/nf-core/scna...

April 11, 2025 at 8:00 PM
scnanoseq nf-co.re/scrnaseq: an nf-core [Nextflow] pipeline for Oxford Nanopore single-cell RNA-sequencing www.biorxiv.org/content/10.1... 🧬🖥️🧪 github.com/nf-core/scna...
Reposted by Jean P. Elbers
Introducing cornetto, an adaptive genome assembly paradigm using @nanoporetech.com adaptive sampling.
- greatly reduces cost per genome assembly
- reference agnostic, so works for non-humans
- assembly just using saliva
- & many more
Relies on 2 excellent software #readfish & #hifiasm.
- greatly reduces cost per genome assembly
- reference agnostic, so works for non-humans
- assembly just using saliva
- & many more
Relies on 2 excellent software #readfish & #hifiasm.

Adaptively integrated sequencing and assembly of near-complete genomes https://www.biorxiv.org/content/10.1101/2025.03.31.646505v1
April 5, 2025 at 4:07 AM
Introducing cornetto, an adaptive genome assembly paradigm using @nanoporetech.com adaptive sampling.
- greatly reduces cost per genome assembly
- reference agnostic, so works for non-humans
- assembly just using saliva
- & many more
Relies on 2 excellent software #readfish & #hifiasm.
- greatly reduces cost per genome assembly
- reference agnostic, so works for non-humans
- assembly just using saliva
- & many more
Relies on 2 excellent software #readfish & #hifiasm.
Reposted by Jean P. Elbers

Congratulations to @imartayan.bsky.social and @curiouscoding.nl whose paper on fast minimizer computation with simd has been accepted to SEA 2025 🙌🏻 www.biorxiv.org/content/10.1...

SimdMinimizers: Computing random minimizers, fast
Motivation Because of the rapidly-growing amount of sequencing data, computing sketches of large textual datasets has become an essential preprocessing task. These sketches are typically much smaller ...
www.biorxiv.org
April 1, 2025 at 8:23 AM
Congratulations to @imartayan.bsky.social and @curiouscoding.nl whose paper on fast minimizer computation with simd has been accepted to SEA 2025 🙌🏻 www.biorxiv.org/content/10.1...
Reposted by Jean P. Elbers

March 10, 2025 at 9:24 PM