



ForliLab
@forlilab.bsky.social
Molecular Modeling & Drug Design. The lab of Stefano Forli at Dept. of Integrative Structural & Computational Biology at @scripps.edu
Home of the AutoDock
https://forlilab.org/
Home of the AutoDock
https://forlilab.org/
Reposted by ForliLab

Explore some of our favourite articles published in Communications Chemistry in 2025:
www.nature.com/collections/...
#ChemSky
www.nature.com/collections/...
#ChemSky

2025 Editors' Highlights
The Editors and Editorial Board Members of Communications Chemistry are pleased to launch a Collection featuring some of their favourite articles published in ...
www.nature.com
January 6, 2026 at 9:54 AM
Explore some of our favourite articles published in Communications Chemistry in 2025:
www.nature.com/collections/...
#ChemSky
www.nature.com/collections/...
#ChemSky
AlphaFold is great, but proteins aren’t single structures.
In our new paper, we introduce AlphaFold-RandomWalk and AlphaFold-Ensemble to sample alternative conformations directly from AlphaFold.
Paper: pubs.acs.org/doi/10.1021/...
and GitHub: github.com/forlilab/pafmd
In our new paper, we introduce AlphaFold-RandomWalk and AlphaFold-Ensemble to sample alternative conformations directly from AlphaFold.
Paper: pubs.acs.org/doi/10.1021/...
and GitHub: github.com/forlilab/pafmd

AlphaFold-RandomWalk and AlphaFold-Ensemble: Sampling Alternative Protein Conformations with Perturbed Versions of AlphaFold
The ability of proteins to adopt multiple conformations is fundamental to their biological function. With the advent of AlphaFold, machine learning (ML)-based methods have extended their capabilities to more broadly sample this intrinsic conformational diversity. However, the extent to which ML approaches can independently generate ensembles of diverse and biologically relevant conformations remains an open question. We sought to tackle this challenge by developing AlphaFold-RandomWalk (AF-RW) and AlphaFold-Ensemble (AF-Ensemble), novel ML-based methods to generate diverse protein conformations. As opposed to traditional approaches which rely on modifying the input multiple sequence alignment, AF-RW systematically adds noise to the weights of the model on a per-target basis, significantly increasing the conformational diversity of predicted models compared to conventional methods. AF-Ensemble takes the complementary approach fine-tuning an ensemble of models to produce diversity from a set of two-state systems. Additionally, both methods were incorporated into an automated, multistage computational pipeline that seeds unbiased molecular dynamics simulations from ML-generated conformations to efficiently sample alternative conformations. When evaluated on a diverse set of ten proteins, our pipeline provided useful, MD-guided hypotheses for determining biologically meaningful alternative conformations. Moreover, simulations seeded from diverse ML-generated conformations provided a reasonable approximation to the free energy landscape of two challenging protein targets, K-Ras and ribose-binding protein. Overall, our work highlights the potential of combining diverse conformations generated by perturbing the weights of AF with molecular dynamics simulations to efficiently probe protein conformational heterogeneity.
pubs.acs.org
January 6, 2026 at 4:58 PM
AlphaFold is great, but proteins aren’t single structures.
In our new paper, we introduce AlphaFold-RandomWalk and AlphaFold-Ensemble to sample alternative conformations directly from AlphaFold.
Paper: pubs.acs.org/doi/10.1021/...
and GitHub: github.com/forlilab/pafmd
In our new paper, we introduce AlphaFold-RandomWalk and AlphaFold-Ensemble to sample alternative conformations directly from AlphaFold.
Paper: pubs.acs.org/doi/10.1021/...
and GitHub: github.com/forlilab/pafmd
Meeko is finally published in JCIM! Check out the paper (pubs.acs.org/doi/full/10....) and GitHub (github.com/forlilab/Meeko) for all your docking preparation needs!

December 11, 2025 at 7:37 PM
Meeko is finally published in JCIM! Check out the paper (pubs.acs.org/doi/full/10....) and GitHub (github.com/forlilab/Meeko) for all your docking preparation needs!
Congratulations to Dr. Ishan Taneja on successfully defending his thesis “Computational Methods for Sampling Alternative Protein Conformations and Modeling Desolvation Upon Protein-Ligand Binding”! He is officially a "Team Forli" graduate!!


November 25, 2025 at 9:25 PM
Congratulations to Dr. Ishan Taneja on successfully defending his thesis “Computational Methods for Sampling Alternative Protein Conformations and Modeling Desolvation Upon Protein-Ligand Binding”! He is officially a "Team Forli" graduate!!
Reposted by ForliLab

Congratulations to eleven Scripps Research scientists named to the global list of #HighlyCited2025 Researchers by @clarivate.com, which recognizes individuals who exemplify research excellence and broad influence across fields including chemistry, microbiology, neuroscience and more.

Scripps Research scientists honored on Clarivate’s Highly Cited Researchers list
ow.ly
November 14, 2025 at 7:20 PM
Congratulations to eleven Scripps Research scientists named to the global list of #HighlyCited2025 Researchers by @clarivate.com, which recognizes individuals who exemplify research excellence and broad influence across fields including chemistry, microbiology, neuroscience and more.
Excited to announce a preprint describing our software package Meeko! Meeko is a Python package that uses RDKit for receptor and ligand preparation, including protonation, bond order, and connectivity and processing of docking results. It is customizable and suitable for high-throughput workflows.
September 18, 2025 at 12:37 AM
Excited to announce a preprint describing our software package Meeko! Meeko is a Python package that uses RDKit for receptor and ligand preparation, including protonation, bond order, and connectivity and processing of docking results. It is customizable and suitable for high-throughput workflows.
Reposted by ForliLab

Excited to share a new preprint from the lab. We show that PTMs like phosphorylation & glycosylation dynamically reshape proteome-wide ligandability in cells, including proteins like KRAS. Great collaboration with the Huang Lab, @forlilab.bsky.social and BMS. www.biorxiv.org/content/10.1...

Post-Translational Modifications Remodel Proteome-Wide Ligandability
Post-translational modifications (PTMs) vastly expand the diversity of human proteome, dynamically reshaping protein activity, interactions, and localization in response to environmental, pharmacologi...
www.biorxiv.org
August 3, 2025 at 2:36 PM
Excited to share a new preprint from the lab. We show that PTMs like phosphorylation & glycosylation dynamically reshape proteome-wide ligandability in cells, including proteins like KRAS. Great collaboration with the Huang Lab, @forlilab.bsky.social and BMS. www.biorxiv.org/content/10.1...
Huge congratulations to Dr. Althea Hansel-Harris on successfully defending her thesis, "Virtual Drug Discovery for Cancer Biology"! Best of luck as you return to medical school at UCSD — we’re excited to see all the amazing things you’ll accomplish! #dr #futuredrdr

April 9, 2025 at 5:34 PM
Huge congratulations to Dr. Althea Hansel-Harris on successfully defending her thesis, "Virtual Drug Discovery for Cancer Biology"! Best of luck as you return to medical school at UCSD — we’re excited to see all the amazing things you’ll accomplish! #dr #futuredrdr
Excited to share our new paper: www.nature.com/articles/s42...! We've compiled the LILAC-DB, a dataset of small molecules bound to membrane proteins at the protein-lipid bilayer interface. (1/5)
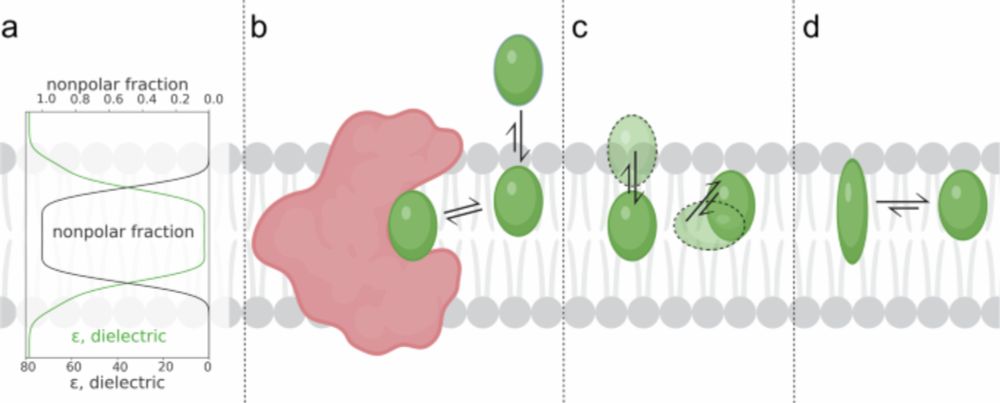
A quantitative analysis of ligand binding at the protein-lipid bilayer interface - Communications Chemistry
Targeting membrane proteins at sites embedded in the lipid bilayer offers the potential to discover ligands for undruggable targets; however, ligand binding at the protein-membrane interface remains u...
www.nature.com
March 26, 2025 at 5:45 PM
Excited to share our new paper: www.nature.com/articles/s42...! We've compiled the LILAC-DB, a dataset of small molecules bound to membrane proteins at the protein-lipid bilayer interface. (1/5)
Reposted by ForliLab

Another big publication from @sijiewang.bsky.social this week. Check out our efforts to use mRNA display to screen for covalent binding inhibitors of Staphylococcus aureus serine hydrolases. pubs.acs.org/articlesonre...
pubs.acs.org
February 27, 2025 at 4:54 PM
Another big publication from @sijiewang.bsky.social this week. Check out our efforts to use mRNA display to screen for covalent binding inhibitors of Staphylococcus aureus serine hydrolases. pubs.acs.org/articlesonre...
A great read for these dire times


February 9, 2025 at 4:15 AM
A great read for these dire times