




F. Kumara Mastrorosa
@fkma.bsky.social
Postdoctoral scholar in the Eichler Lab at UW Genome Sciences. Interested in Mendelian disorders, long-read sequencing and structural variations 🇮🇹 🇪🇺
Reposted by F. Kumara Mastrorosa
Thank you Dr. Danny Miller @danrdanny.bsky.social for hosting, and fantastic job Dr. Mastrorosa @fkma.bsky.social!
@uwgenome.bsky.social
brotmanbaty.org/news/long-re...
@uwgenome.bsky.social
brotmanbaty.org/news/long-re...

September 25, 2025 at 5:19 PM
Thank you Dr. Danny Miller @danrdanny.bsky.social for hosting, and fantastic job Dr. Mastrorosa @fkma.bsky.social!
@uwgenome.bsky.social
brotmanbaty.org/news/long-re...
@uwgenome.bsky.social
brotmanbaty.org/news/long-re...
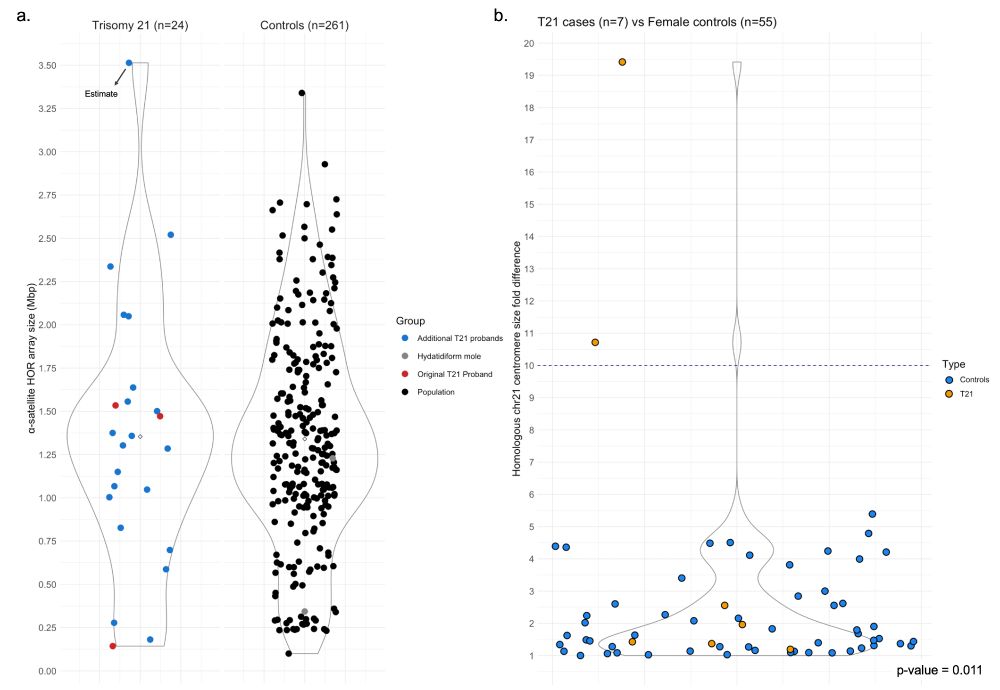
We updated our preprint describing chr21 centromere genetics/epigenetics in families with Down syndrome and general population! We found transgenerational methylation changes in a subset of families and that centromere size asymmetry is exclusive to T21! www.biorxiv.org/content/10.1...
July 18, 2025 at 5:47 PM
We updated our preprint describing chr21 centromere genetics/epigenetics in families with Down syndrome and general population! We found transgenerational methylation changes in a subset of families and that centromere size asymmetry is exclusive to T21! www.biorxiv.org/content/10.1...
A deep analysis of the 22q11.2 locus in the human population and affected individuals studying the mechanisms and architectures that predispose to complex rearrangements!
July 9, 2025 at 9:27 PM
A deep analysis of the 22q11.2 locus in the human population and affected individuals studying the mechanisms and architectures that predispose to complex rearrangements!
I am very happy to have contributed to the publication of the first high-quality, near-complete Middle Eastern genomes. These data will help population studies, disease gene discovery, and increase population representation in genomic datasets!

📢 ONLINE @natgenet.nature.com
📰Near-complete Middle Eastern genomes refine autozygosity and enhance disease-causing and population-specific variant discovery.
By Mohammadmersad Ghorbani, Younes Mokrab and colleagues.
⬇️
www.nature.com/articles/s41...
📰Near-complete Middle Eastern genomes refine autozygosity and enhance disease-causing and population-specific variant discovery.
By Mohammadmersad Ghorbani, Younes Mokrab and colleagues.
⬇️
www.nature.com/articles/s41...

Near-complete Middle Eastern genomes refine autozygosity and enhance disease-causing and population-specific variant discovery - Nature Genetics
Generation and analysis of high-quality, genome assemblies from Middle Eastern trios demonstrate the utility of ancestry-matched data and assembly-based variation analysis.
www.nature.com
June 27, 2025 at 5:50 PM
I am very happy to have contributed to the publication of the first high-quality, near-complete Middle Eastern genomes. These data will help population studies, disease gene discovery, and increase population representation in genomic datasets!
The most accurate way so far to study transmission, de novo variants, and recombination!
Our Nature paper (rdcu.be/ei1NM) deep sequencing a 4-generation, 28-member family using multiple sequencing technologies to study transmission of all classes of genetic variation is out! @uwgenome.bsky.social @hhmi.org @pacbio.bsky.social @utah.edu
April 28, 2025 at 6:29 PM
The most accurate way so far to study transmission, de novo variants, and recombination!
Reposted by F. Kumara Mastrorosa


We recently published CDR-Finder, a tool to study hypomethylated regions in centromeres (academic.oup.com/bioinformati...). The latest update allows to use the tool to study any region of the genome! This will be useful for neocentromeres, promoters and any methylation variable locus!

Identification and annotation of centromeric hypomethylated regions with CDR-Finder
AbstractMotivation. Centromeres are chromosomal regions historically understudied with sequencing technologies due to their repetitive nature and short-rea
academic.oup.com
February 5, 2025 at 5:22 PM
We recently published CDR-Finder, a tool to study hypomethylated regions in centromeres (academic.oup.com/bioinformati...). The latest update allows to use the tool to study any region of the genome! This will be useful for neocentromeres, promoters and any methylation variable locus!