




Aaron Kwok
@aaronkwc.bsky.social
PhD student at Bioinformatics and Cellular Genomics group @SVIResearch
Pinned
Aaron Kwok
@aaronkwc.bsky.social
· Dec 11
Have you been thinking hard about statistical modelling of scATAC-seq data? (No.)
Luckily for you, @aaronkwc.bsky.social has!
Aaron will help you grok:
What's going on?
What is TF-IDF?
Is there really single-cell level chromatin information?
Check it out 👇
www.biorxiv.org/content/10.1...
🧪🧬💻
Luckily for you, @aaronkwc.bsky.social has!
Aaron will help you grok:
What's going on?
What is TF-IDF?
Is there really single-cell level chromatin information?
Check it out 👇
www.biorxiv.org/content/10.1...
🧪🧬💻

Going beyond cell clustering and feature aggregation: Is there single cell level information in single-cell ATAC-seq data?
Single-cell Assay for Transposase Accessible Chromatin with sequencing (scATAC-seq) has become a widely used method for investigating chromatin accessibility at single-cell resolution. However, the re...
www.biorxiv.org
Many thanks to my supervisors @davisjmcc.bsky.social and Heejung who encouraged me to write this piece up! It is not a style of writing that I am the most familiar with so it took a lot of experimenting and also valuable feedback from @jeffreypullin.bsky.social on some earlier versions
Reposted by Aaron Kwok

The McCarthy lab @davisjmcc.bsky.social is very excited to be in beautiful Adelaide for the 10th ABACBS conference. Look at our happy faces @aaronkwc.bsky.social @ameliadunstone #ABACBS2025

November 24, 2025 at 10:55 PM
The McCarthy lab @davisjmcc.bsky.social is very excited to be in beautiful Adelaide for the 10th ABACBS conference. Look at our happy faces @aaronkwc.bsky.social @ameliadunstone #ABACBS2025
Reposted by Aaron Kwok

Great new work led by Aaron Kwok from @davisjmcc.bsky.social’s group. A tool to “denoise” contaminating transcripts from image based spatial data.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...

Denoising image-based spatial transcriptomics data with DenoIST
Image-based spatial transcriptomics (IST) technologies provide unprecedented resolution of gene expression in tissue sections, but suffer from contamination of cells' gene expression profiles due to i...
www.biorxiv.org
November 15, 2025 at 1:31 PM
Great new work led by Aaron Kwok from @davisjmcc.bsky.social’s group. A tool to “denoise” contaminating transcripts from image based spatial data.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
Reposted by Aaron Kwok

Very excited to share new work from my PhD on a new software package for eQTL mapping: quasar. The quasar software package is a C++ program designed to provide a flexible and efficient eQTL mapping. www.medrxiv.org/content/10.1...

Flexible and efficient count-distribution and mixed-model methods for eQTL mapping with quasar
Identifying genetic variants that affect gene expression, expression quantitative trait loci (eQTLs), is a major focus of modern genomics. Today, various methods exist for eQTL mapping, each using dif...
www.medrxiv.org
July 22, 2025 at 10:15 AM
Very excited to share new work from my PhD on a new software package for eQTL mapping: quasar. The quasar software package is a C++ program designed to provide a flexible and efficient eQTL mapping. www.medrxiv.org/content/10.1...
Reposted by Aaron Kwok

Here it is! Bonsai. Now there is really no more excuse for using t-SNE/UMAP. Bonsai not only makes cool pictures of your data. It actually rigorously preserves its structure. No tunable parameters. Incredible work by @dhdegroot.bsky.social.
I'm so excited about this!
www.biorxiv.org/content/10.1...
I'm so excited about this!
www.biorxiv.org/content/10.1...

Bonsai: Tree representations for distortion-free visualization and exploratory analysis of single-cell omics data
Single-cell omics methods promise to revolutionize our understanding of gene regulatory processes during cell differentiation, but analysis of such data continues to pose a major challenge. Apart from...
www.biorxiv.org
May 9, 2025 at 10:49 AM
Here it is! Bonsai. Now there is really no more excuse for using t-SNE/UMAP. Bonsai not only makes cool pictures of your data. It actually rigorously preserves its structure. No tunable parameters. Incredible work by @dhdegroot.bsky.social.
I'm so excited about this!
www.biorxiv.org/content/10.1...
I'm so excited about this!
www.biorxiv.org/content/10.1...
Reposted by Aaron Kwok
Excited to share Ruqian has developed a package for this transcript based niche analysis. Check out the preprint and the package! This is a flexible approach and we're working on extending this for other spatial analyses!
@davisjmcc.bsky.social
www.biorxiv.org/content/10.1...
@davisjmcc.bsky.social
www.biorxiv.org/content/10.1...
March 26, 2025 at 6:04 PM
Excited to share Ruqian has developed a package for this transcript based niche analysis. Check out the preprint and the package! This is a flexible approach and we're working on extending this for other spatial analyses!
@davisjmcc.bsky.social
www.biorxiv.org/content/10.1...
@davisjmcc.bsky.social
www.biorxiv.org/content/10.1...
Reposted by Aaron Kwok

How to do differential expression with scRNAseq data? State of the art is "pseudo-bulk" analysis with RNA-seq methods like edgeR or DESeq2, where "cell type" is encoded as discrete categories. Biologically, discrete categories are not always the most appropriate concept.(1/3)
doi.org/10.1038/s415...
doi.org/10.1038/s415...
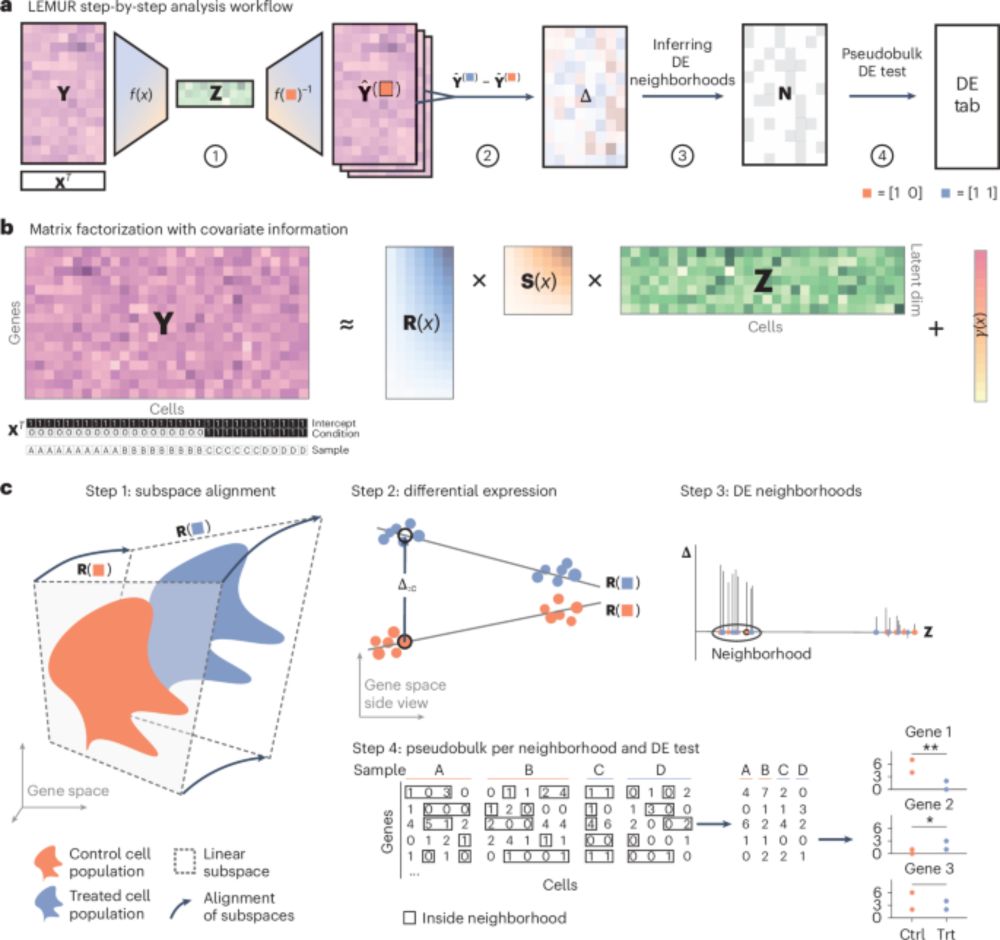
Analysis of multi-condition single-cell data with latent embedding multivariate regression - Nature Genetics
Latent embedding multivariate regression models multi-condition single-cell RNA-seq using a continuous latent space, enabling data integration, per-cell gene expression prediction and clustering-free ...
doi.org
January 3, 2025 at 7:18 PM
How to do differential expression with scRNAseq data? State of the art is "pseudo-bulk" analysis with RNA-seq methods like edgeR or DESeq2, where "cell type" is encoded as discrete categories. Biologically, discrete categories are not always the most appropriate concept.(1/3)
doi.org/10.1038/s415...
doi.org/10.1038/s415...
Reposted by Aaron Kwok

Reposted by Aaron Kwok

What do GWAS and rare variant burden tests discover, and why?
Do these studies find the most IMPORTANT genes? If not, how DO they rank genes?
Here we present a surprising result: these studies actually test for SPECIFICITY! A 🧵on what this means... (🧪🧬)
www.biorxiv.org/content/10.1...
Do these studies find the most IMPORTANT genes? If not, how DO they rank genes?
Here we present a surprising result: these studies actually test for SPECIFICITY! A 🧵on what this means... (🧪🧬)
www.biorxiv.org/content/10.1...

Specificity, length, and luck: How genes are prioritized by rare and common variant association studies
Standard genome-wide association studies (GWAS) and rare variant burden tests are essential tools for identifying trait-relevant genes. Although these methods are conceptually similar, we show by anal...
www.biorxiv.org
December 17, 2024 at 7:05 AM
What do GWAS and rare variant burden tests discover, and why?
Do these studies find the most IMPORTANT genes? If not, how DO they rank genes?
Here we present a surprising result: these studies actually test for SPECIFICITY! A 🧵on what this means... (🧪🧬)
www.biorxiv.org/content/10.1...
Do these studies find the most IMPORTANT genes? If not, how DO they rank genes?
Here we present a surprising result: these studies actually test for SPECIFICITY! A 🧵on what this means... (🧪🧬)
www.biorxiv.org/content/10.1...
Many thanks to my supervisors @davisjmcc.bsky.social and Heejung who encouraged me to write this piece up! It is not a style of writing that I am the most familiar with so it took a lot of experimenting and also valuable feedback from @jeffreypullin.bsky.social on some earlier versions
Have you been thinking hard about statistical modelling of scATAC-seq data? (No.)
Luckily for you, @aaronkwc.bsky.social has!
Aaron will help you grok:
What's going on?
What is TF-IDF?
Is there really single-cell level chromatin information?
Check it out 👇
www.biorxiv.org/content/10.1...
🧪🧬💻
Luckily for you, @aaronkwc.bsky.social has!
Aaron will help you grok:
What's going on?
What is TF-IDF?
Is there really single-cell level chromatin information?
Check it out 👇
www.biorxiv.org/content/10.1...
🧪🧬💻
Going beyond cell clustering and feature aggregation: Is there single cell level information in single-cell ATAC-seq data?
Single-cell Assay for Transposase Accessible Chromatin with sequencing (scATAC-seq) has become a widely used method for investigating chromatin accessibility at single-cell resolution. However, the re...
www.biorxiv.org
December 11, 2024 at 1:21 AM
Many thanks to my supervisors @davisjmcc.bsky.social and Heejung who encouraged me to write this piece up! It is not a style of writing that I am the most familiar with so it took a lot of experimenting and also valuable feedback from @jeffreypullin.bsky.social on some earlier versions
Reposted by Aaron Kwok

New work! Wherein we (as in, the AVE ODIC working group) looked at clinical variant classification across genetic ancestry groups in gnomAD and AoU and what we found... is exactly what you might expect, after decades of Eurocentric research. But we also show there's a better way forward! 🧬🖥️

Defining and Reducing Variant Classification Disparities https://www.medrxiv.org/content/10.1101/2024.04.11.24305690v1

Defining and Reducing Variant Classification Disparities https://www.medrxiv.org/content/10.1101/2024.04.11.24305690v1
Background: Multiplexed Assays of Variant Effects (MAVEs) can test all possible single variants in a
www.medrxiv.org
April 18, 2024 at 11:07 PM
New work! Wherein we (as in, the AVE ODIC working group) looked at clinical variant classification across genetic ancestry groups in gnomAD and AoU and what we found... is exactly what you might expect, after decades of Eurocentric research. But we also show there's a better way forward! 🧬🖥️
Reposted by Aaron Kwok

Have you been thinking hard about statistical modelling of scATAC-seq data? (No.)
Luckily for you, @aaronkwc.bsky.social has!
Aaron will help you grok:
What's going on?
What is TF-IDF?
Is there really single-cell level chromatin information?
Check it out 👇
www.biorxiv.org/content/10.1...
🧪🧬💻
Luckily for you, @aaronkwc.bsky.social has!
Aaron will help you grok:
What's going on?
What is TF-IDF?
Is there really single-cell level chromatin information?
Check it out 👇
www.biorxiv.org/content/10.1...
🧪🧬💻
Going beyond cell clustering and feature aggregation: Is there single cell level information in single-cell ATAC-seq data?
Single-cell Assay for Transposase Accessible Chromatin with sequencing (scATAC-seq) has become a widely used method for investigating chromatin accessibility at single-cell resolution. However, the re...
www.biorxiv.org
December 10, 2024 at 6:37 AM
Have you been thinking hard about statistical modelling of scATAC-seq data? (No.)
Luckily for you, @aaronkwc.bsky.social has!
Aaron will help you grok:
What's going on?
What is TF-IDF?
Is there really single-cell level chromatin information?
Check it out 👇
www.biorxiv.org/content/10.1...
🧪🧬💻
Luckily for you, @aaronkwc.bsky.social has!
Aaron will help you grok:
What's going on?
What is TF-IDF?
Is there really single-cell level chromatin information?
Check it out 👇
www.biorxiv.org/content/10.1...
🧪🧬💻