



Paul Robustelli
@paulrobustelli.bsky.social
Assistant Professor at Dartmouth College
Computational Biophysics / Disordered Proteins / Molecular Recognition
Computational Biophysics / Disordered Proteins / Molecular Recognition
Today Writhe, our favorite knot theory descriptor for characterizing IDP dynamics, is the star of the lab pumpkin for the annual department carving contest.
If you can make sense of the design— I’ll buy you a coffee, bao bun, or wheatgrass shot at the next conference. No substitutions.
If you can make sense of the design— I’ll buy you a coffee, bao bun, or wheatgrass shot at the next conference. No substitutions.



October 23, 2025 at 1:05 AM
Today Writhe, our favorite knot theory descriptor for characterizing IDP dynamics, is the star of the lab pumpkin for the annual department carving contest.
If you can make sense of the design— I’ll buy you a coffee, bao bun, or wheatgrass shot at the next conference. No substitutions.
If you can make sense of the design— I’ll buy you a coffee, bao bun, or wheatgrass shot at the next conference. No substitutions.
Fun day at @dartmouthchem.bsky.social
serving buldak ramen and explaining the biophysics and structural biology of heat and capsaicin activation of the ion channels to celebrate @acs.org #NationalChemistryWeek.
serving buldak ramen and explaining the biophysics and structural biology of heat and capsaicin activation of the ion channels to celebrate @acs.org #NationalChemistryWeek.

October 20, 2025 at 6:52 PM
Fun day at @dartmouthchem.bsky.social
serving buldak ramen and explaining the biophysics and structural biology of heat and capsaicin activation of the ion channels to celebrate @acs.org #NationalChemistryWeek.
serving buldak ramen and explaining the biophysics and structural biology of heat and capsaicin activation of the ion channels to celebrate @acs.org #NationalChemistryWeek.
Also, take note of our 15 (!) exp. refined all-atom IDP ensembles deposited in the protein ensemble database:
proteinensemble.org/entries/PED0...
If you think you have good method / force field for generating IDP ensembles, you can benchmark your agreement with exp. against these ensembles.
proteinensemble.org/entries/PED0...
If you think you have good method / force field for generating IDP ensembles, you can benchmark your agreement with exp. against these ensembles.

October 10, 2025 at 3:47 PM
Also, take note of our 15 (!) exp. refined all-atom IDP ensembles deposited in the protein ensemble database:
proteinensemble.org/entries/PED0...
If you think you have good method / force field for generating IDP ensembles, you can benchmark your agreement with exp. against these ensembles.
proteinensemble.org/entries/PED0...
If you think you have good method / force field for generating IDP ensembles, you can benchmark your agreement with exp. against these ensembles.
While they're only rarely populated at the same time - these multisite binding mode give us a better understanding of how a network of hydrogen bond donors and acceptors confer pronounced affinity to residues 7_CATQRLANFLV_17.

September 27, 2025 at 1:45 PM
While they're only rarely populated at the same time - these multisite binding mode give us a better understanding of how a network of hydrogen bond donors and acceptors confer pronounced affinity to residues 7_CATQRLANFLV_17.
To get more insight into the diversity of binding modes and search for more structured modes - we looked at binding poses where at least 15 residues of hIAPP were in contact with each ligand. This represented 12.9% of bound frames for YX-I-1 but only 3.9% of YX-A-1.


September 27, 2025 at 1:45 PM
To get more insight into the diversity of binding modes and search for more structured modes - we looked at binding poses where at least 15 residues of hIAPP were in contact with each ligand. This represented 12.9% of bound frames for YX-I-1 but only 3.9% of YX-A-1.
Interestingly, the exposed regions of YX-A-1 are quite hydrophobic (cylcohexane and benzene) - and we think this could be part of how it accelerates aggregation into higher order oligomers and protofilaments
September 27, 2025 at 1:45 PM
Interestingly, the exposed regions of YX-A-1 are quite hydrophobic (cylcohexane and benzene) - and we think this could be part of how it accelerates aggregation into higher order oligomers and protofilaments
We see that each ligand has moieties that are consistently buried and others that are consistently exposed across binding modes. We think that buried moieties might confer monomer affinity - while exposed moieties could affect rates of oligomerization into higher order species.


September 27, 2025 at 1:45 PM
We see that each ligand has moieties that are consistently buried and others that are consistently exposed across binding modes. We think that buried moieties might confer monomer affinity - while exposed moieties could affect rates of oligomerization into higher order species.
Comparing populations of intermolecular interactions we found something unique about this pair: the largest difference is elevated populations of hydrogen bonds with YX-I-1, not increased populations of aromatic stacking interactions -which we usually see in tighter IDP ligands

September 27, 2025 at 1:45 PM
Comparing populations of intermolecular interactions we found something unique about this pair: the largest difference is elevated populations of hydrogen bonds with YX-I-1, not increased populations of aromatic stacking interactions -which we usually see in tighter IDP ligands
We have a detailed comparison of contact profiles and helicity changes with NMR CSPs from in the SI. We don't see perfect agreement, but observe that the average magnitude of CSPs correlate pretty well with average contact populations and changes in helicity upon and binding.


September 27, 2025 at 1:45 PM
We have a detailed comparison of contact profiles and helicity changes with NMR CSPs from in the SI. We don't see perfect agreement, but observe that the average magnitude of CSPs correlate pretty well with average contact populations and changes in helicity upon and binding.
In ligand binding simulations, we see heterogenous ensembles of binding modes. We see YX-I-1 is a tighter binder than YX-A-1, and produces larger conformational changes upon binding, consistent with larger NMR chemical shift perturbations (CSPs) and other assays from @radford-lab.bsky.social

September 27, 2025 at 1:45 PM
In ligand binding simulations, we see heterogenous ensembles of binding modes. We see YX-I-1 is a tighter binder than YX-A-1, and produces larger conformational changes upon binding, consistent with larger NMR chemical shift perturbations (CSPs) and other assays from @radford-lab.bsky.social
Michelle had an awesome idea to use matrices of circuit topology assignments from work by @alirezamashaghi.bsky.social (pubs.acs.org/doi/full/10....) for dimensionality reduction to project all our apo and holo ensembles onto latent space reflecting the topological similarity of conformations.


September 27, 2025 at 1:45 PM
Michelle had an awesome idea to use matrices of circuit topology assignments from work by @alirezamashaghi.bsky.social (pubs.acs.org/doi/full/10....) for dimensionality reduction to project all our apo and holo ensembles onto latent space reflecting the topological similarity of conformations.
Our WT hIAPP ensemble is in good agreement with NMR chemical shifts, showing us we have a good force field (a99SB-disp) for this system. S20G introduces a central hinge that increases populations of intramolecular contact and beta-sheets bewteen residues in hIAPP fibril cores


September 27, 2025 at 1:45 PM
Our WT hIAPP ensemble is in good agreement with NMR chemical shifts, showing us we have a good force field (a99SB-disp) for this system. S20G introduces a central hinge that increases populations of intramolecular contact and beta-sheets bewteen residues in hIAPP fibril cores
We used 400us of enhanced sampling (REST2) all-atom MD to characterize the conformational ensembles of wild-type (WT) hIAPP, and the S20G variant (which accelerates aggregation and is associated with early-onset T2D) and characterize their interactions with these ligands.
September 27, 2025 at 1:45 PM
We used 400us of enhanced sampling (REST2) all-atom MD to characterize the conformational ensembles of wild-type (WT) hIAPP, and the S20G variant (which accelerates aggregation and is associated with early-onset T2D) and characterize their interactions with these ligands.
They found molecules that inhibit (YX-I-1) and accelerate (YX-A-1) hIAPP aggregation.
YX-I-1, which was found to bind monomer by NMR, SPR, and mass-spec could be a lead for developing T2D therapies. Kinetics assays show YX-A-1 mainly interacts with higher order oligomers.
YX-I-1, which was found to bind monomer by NMR, SPR, and mass-spec could be a lead for developing T2D therapies. Kinetics assays show YX-A-1 mainly interacts with higher order oligomers.

September 27, 2025 at 1:45 PM
They found molecules that inhibit (YX-I-1) and accelerate (YX-A-1) hIAPP aggregation.
YX-I-1, which was found to bind monomer by NMR, SPR, and mass-spec could be a lead for developing T2D therapies. Kinetics assays show YX-A-1 mainly interacts with higher order oligomers.
YX-I-1, which was found to bind monomer by NMR, SPR, and mass-spec could be a lead for developing T2D therapies. Kinetics assays show YX-A-1 mainly interacts with higher order oligomers.
Aggregation and amyloid formation of the disordered protein human islet amyloid polypeptide (hIAPP) is associated with type-2-diabetes (T2D).
Recently, @radford-lab.bsky.social ran a screen of 1500 small molecules to find hIAPP binders.
nature.com/articles/s41...
Recently, @radford-lab.bsky.social ran a screen of 1500 small molecules to find hIAPP binders.
nature.com/articles/s41...

September 27, 2025 at 1:45 PM
Aggregation and amyloid formation of the disordered protein human islet amyloid polypeptide (hIAPP) is associated with type-2-diabetes (T2D).
Recently, @radford-lab.bsky.social ran a screen of 1500 small molecules to find hIAPP binders.
nature.com/articles/s41...
Recently, @radford-lab.bsky.social ran a screen of 1500 small molecules to find hIAPP binders.
nature.com/articles/s41...
Excited to share a new preprint:
"Monomer binding modes of small molecules that modulate the kinetics of hIAPP amyloid formation"
by graduate student Michelle Garcia together with post-doc Korey Reid.
Paper:
www.biorxiv.org/content/10.1...
Code + Ensembles: github.com/paulrobustel...
"Monomer binding modes of small molecules that modulate the kinetics of hIAPP amyloid formation"
by graduate student Michelle Garcia together with post-doc Korey Reid.
Paper:
www.biorxiv.org/content/10.1...
Code + Ensembles: github.com/paulrobustel...
September 27, 2025 at 1:45 PM
Excited to share a new preprint:
"Monomer binding modes of small molecules that modulate the kinetics of hIAPP amyloid formation"
by graduate student Michelle Garcia together with post-doc Korey Reid.
Paper:
www.biorxiv.org/content/10.1...
Code + Ensembles: github.com/paulrobustel...
"Monomer binding modes of small molecules that modulate the kinetics of hIAPP amyloid formation"
by graduate student Michelle Garcia together with post-doc Korey Reid.
Paper:
www.biorxiv.org/content/10.1...
Code + Ensembles: github.com/paulrobustel...
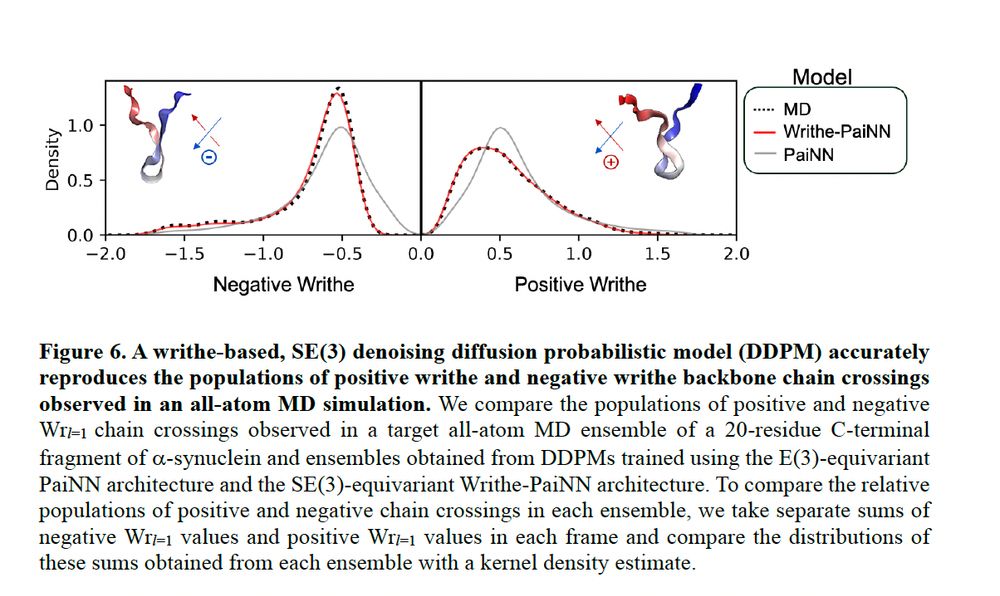
As a proof-of-principle, we showed that if you train DDPMs with Writhe-PaiNN on a single long timescale MD trajectory, you can accurately described the populations of chiral chain crossings seen in that simulations, whereas a DDPM trained with PaiNN can't distinguish their populations.
April 30, 2025 at 5:45 PM
As a proof-of-principle, we showed that if you train DDPMs with Writhe-PaiNN on a single long timescale MD trajectory, you can accurately described the populations of chiral chain crossings seen in that simulations, whereas a DDPM trained with PaiNN can't distinguish their populations.
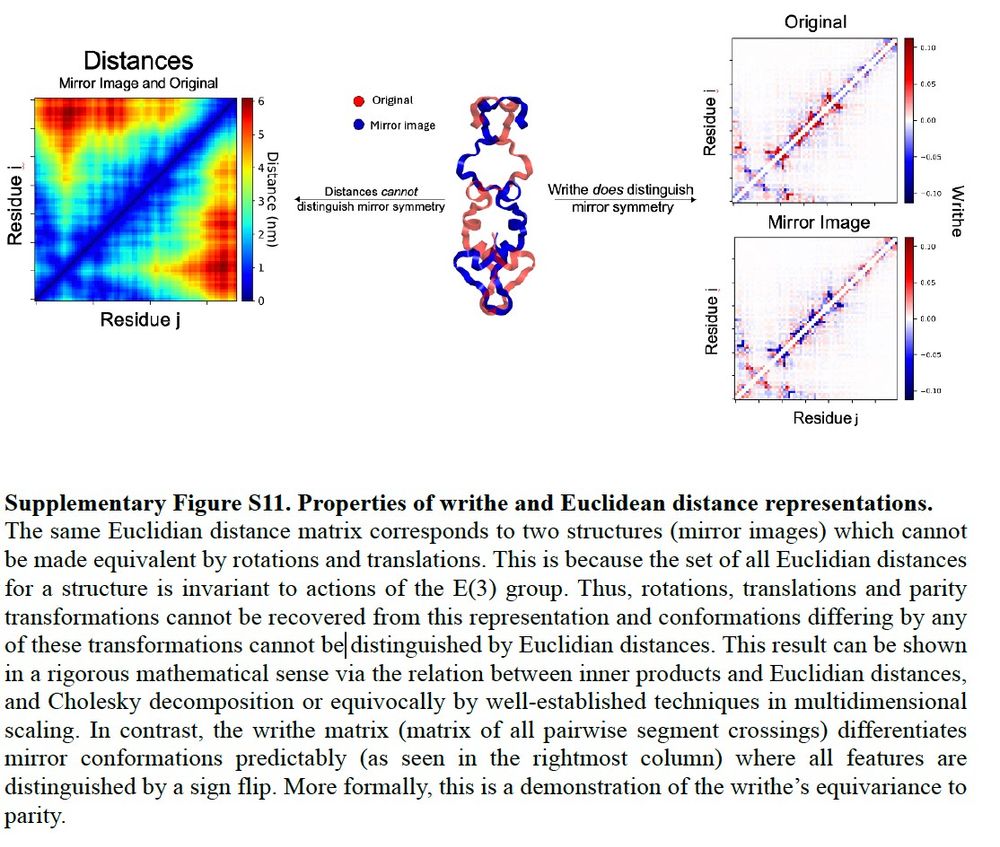
But there's more. Tommy loves neural networks (NNs) and generative models, which need to be trained using NN architectures that conserve or exclude certain geometric and symmetry properties of coordinate data. Tommy saw that if you take a mirror reflection of a protein conformation the...
April 30, 2025 at 5:45 PM
But there's more. Tommy loves neural networks (NNs) and generative models, which need to be trained using NN architectures that conserve or exclude certain geometric and symmetry properties of coordinate data. Tommy saw that if you take a mirror reflection of a protein conformation the...
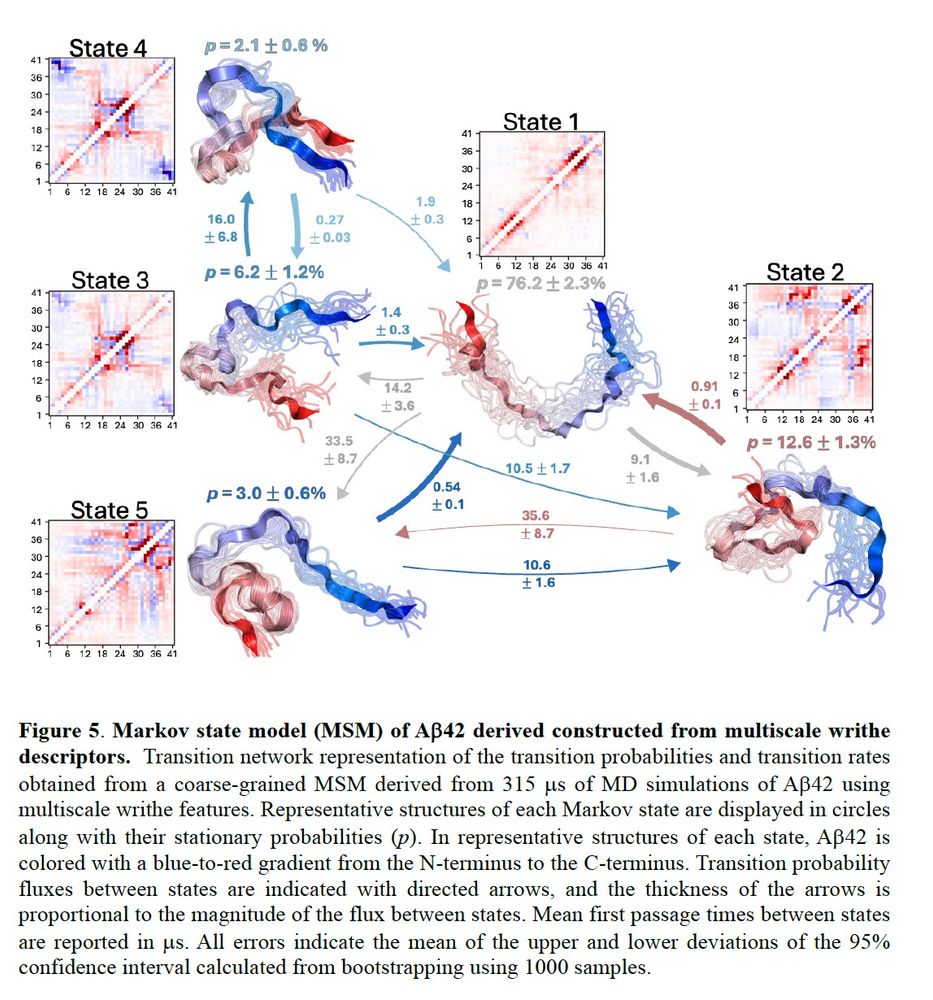
Now we can take this MSM workflow and build a very nice 5-macrostate MSM from 315us of Abeta42 MD simulations from @tlhr.bsky.social and Vendruscolo lab coworkers (www.nature.com/articles/s43...), without using any of the elegant ML tricks of their original paper.
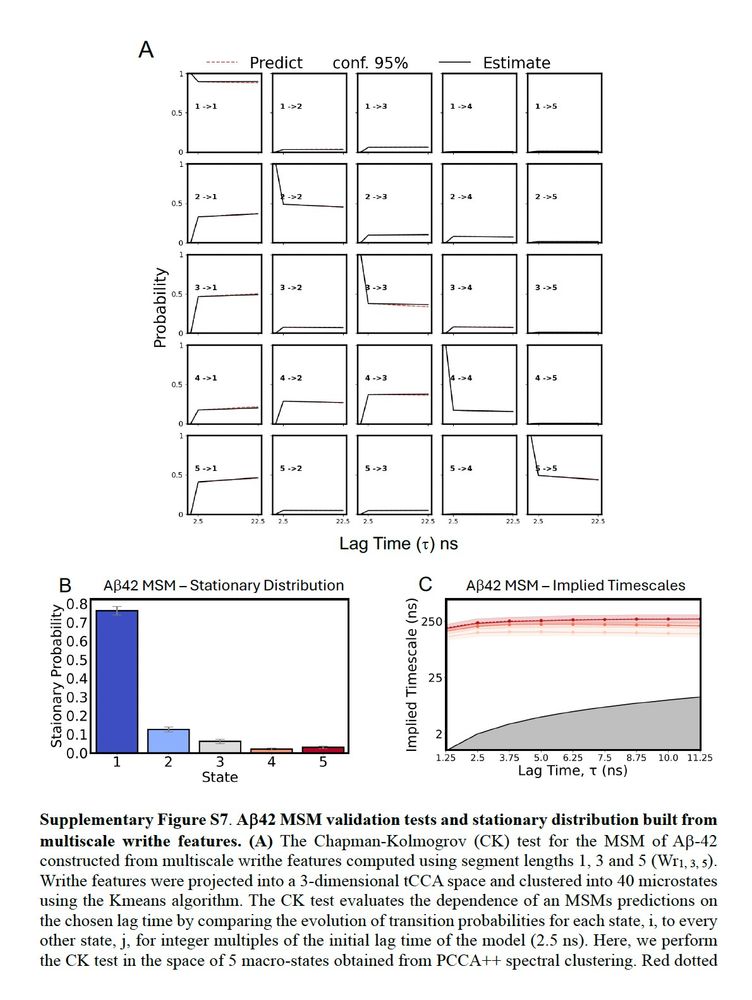
April 30, 2025 at 5:45 PM
Now we can take this MSM workflow and build a very nice 5-macrostate MSM from 315us of Abeta42 MD simulations from @tlhr.bsky.social and Vendruscolo lab coworkers (www.nature.com/articles/s43...), without using any of the elegant ML tricks of their original paper.
If we build an MSM from these projections, we see that the largest implied time scale (ITS) of the writhe MSM converges to a substantially larger value than the largest ITS of the inter-residue distance MSM.
Lucky break? Nope! We see this for all our IDP simulations scanning many MSM parameters.
Lucky break? Nope! We see this for all our IDP simulations scanning many MSM parameters.
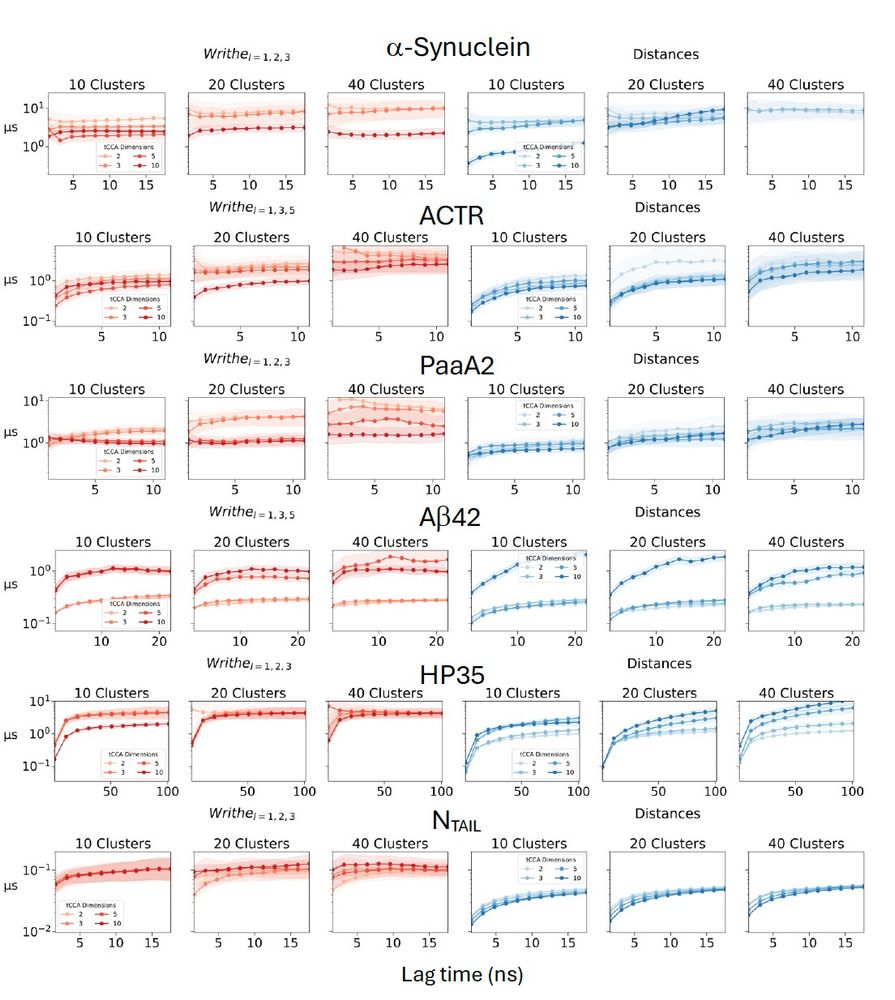

April 30, 2025 at 5:45 PM
If we build an MSM from these projections, we see that the largest implied time scale (ITS) of the writhe MSM converges to a substantially larger value than the largest ITS of the inter-residue distance MSM.
Lucky break? Nope! We see this for all our IDP simulations scanning many MSM parameters.
Lucky break? Nope! We see this for all our IDP simulations scanning many MSM parameters.
Now let's look at how many discrete conformational states we identify from writhe tCCA projections (or reaction coordinates) and distance tCCA projections for a 30us IDP simulation. Here, we see that writhe computed at single segment length (no cheating) identifies more states than distances.
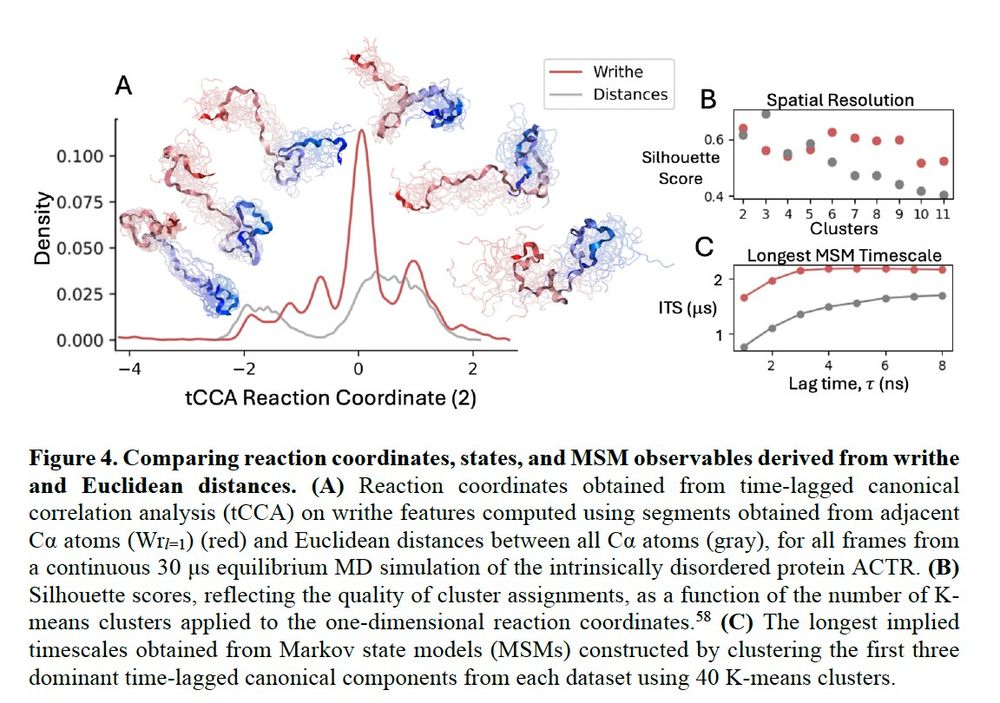
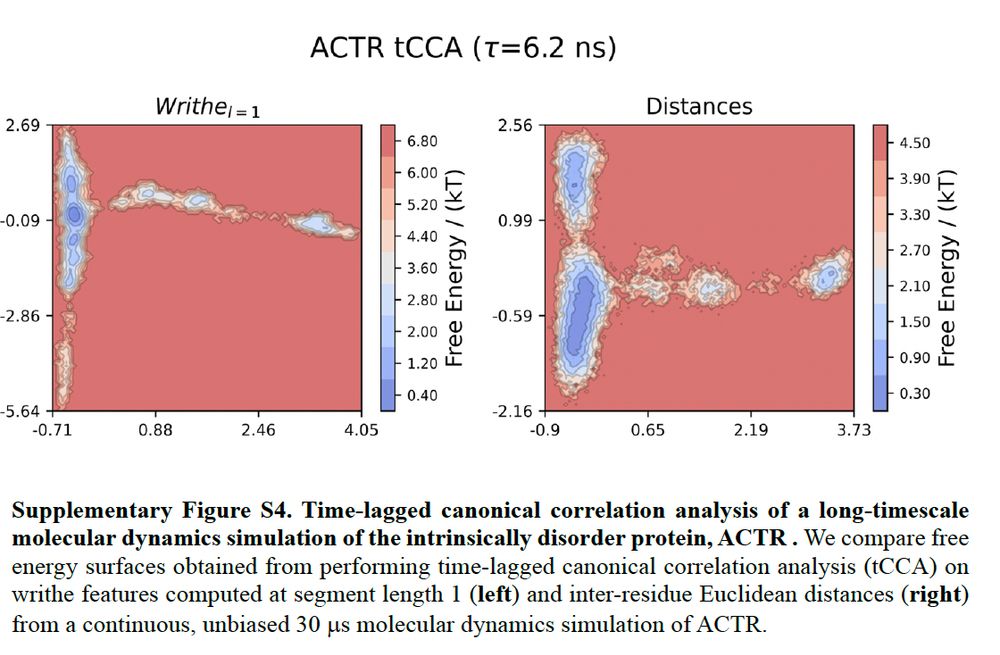
April 30, 2025 at 5:45 PM
Now let's look at how many discrete conformational states we identify from writhe tCCA projections (or reaction coordinates) and distance tCCA projections for a 30us IDP simulation. Here, we see that writhe computed at single segment length (no cheating) identifies more states than distances.
..the slowest motions in equilibrium MD simulations of 5 IDPs and a fast folding protein. We're going to perform tCCA using writhe from different segment lengths, and compute the kinetic variance (aka VAMP-2 score) of the slowest modes at different lag times and compare to interresidue distances
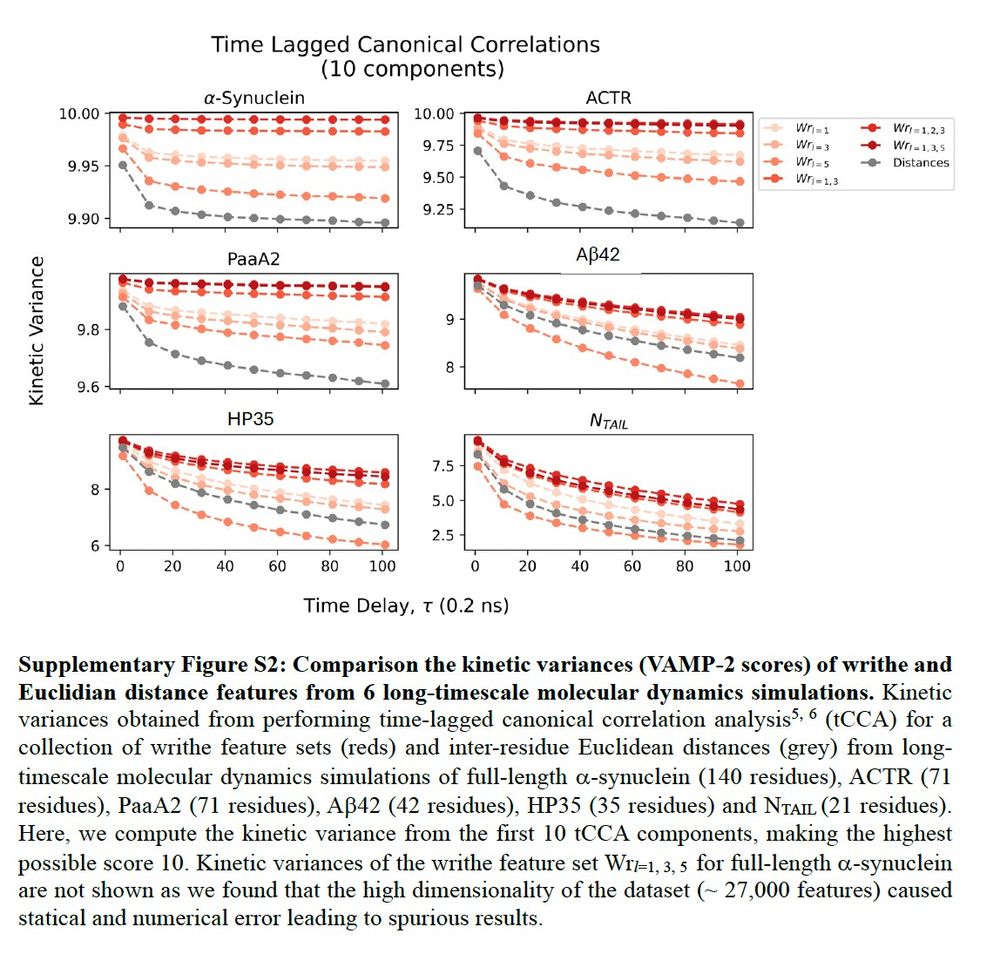
April 30, 2025 at 5:45 PM
..the slowest motions in equilibrium MD simulations of 5 IDPs and a fast folding protein. We're going to perform tCCA using writhe from different segment lengths, and compute the kinetic variance (aka VAMP-2 score) of the slowest modes at different lag times and compare to interresidue distances
Polymers (and IDPs) have different timescale fluctuations at different length scales. We compare writhe matrices computed with different segment lengths for a folded conformation of HP35, and show that they are sensitive to different structural features. We analyze a folding trajectory of HP35...
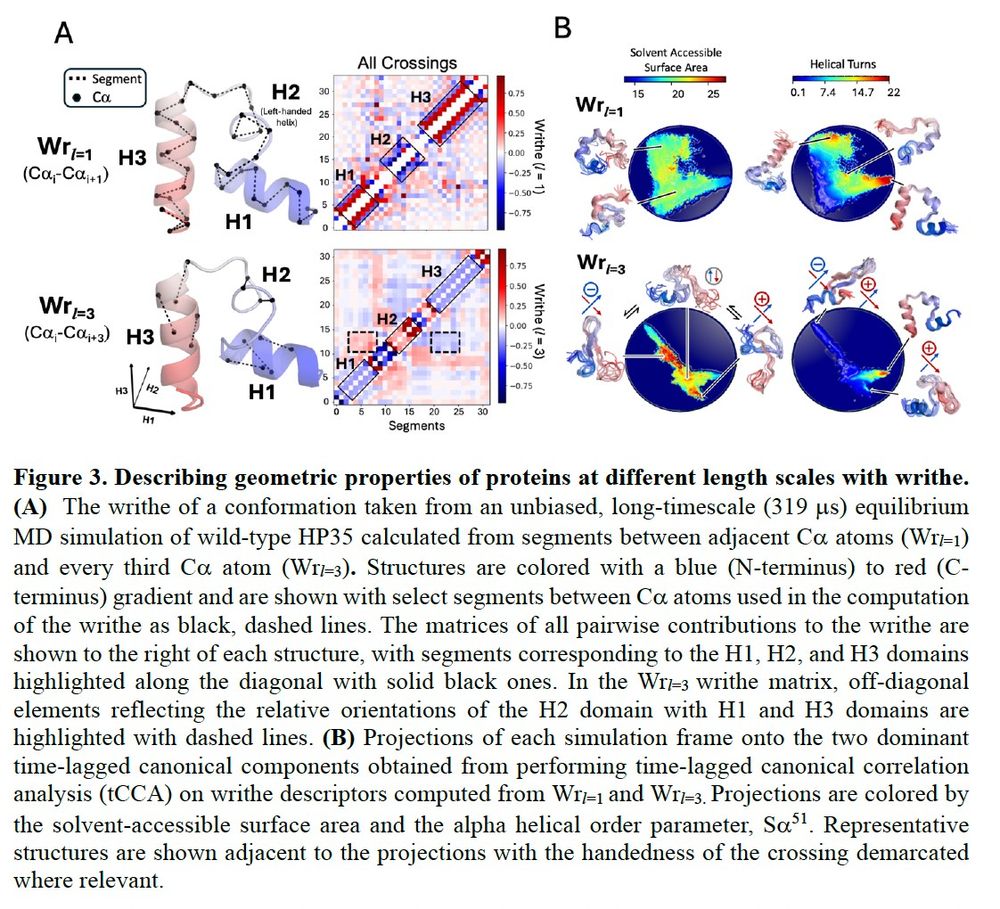
April 30, 2025 at 5:45 PM
Polymers (and IDPs) have different timescale fluctuations at different length scales. We compare writhe matrices computed with different segment lengths for a folded conformation of HP35, and show that they are sensitive to different structural features. We analyze a folding trajectory of HP35...
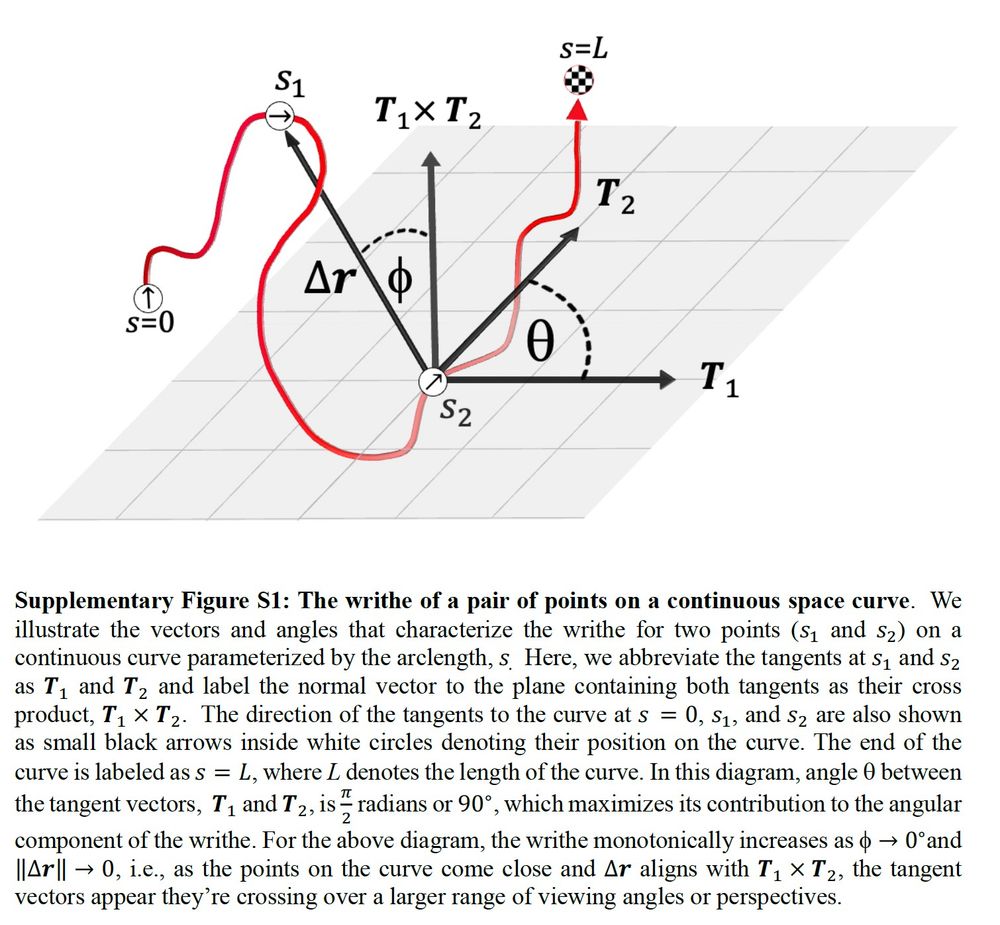
Here is an illustration of how to compute writhe in a geometric fashion. You can project the apparent crossings of two segments to define a spherical quadrangle, and use this the accurately calculate writhe. Tommy wrote a python package to do this real fast
github.com/paulrobustel...
github.com/paulrobustel...
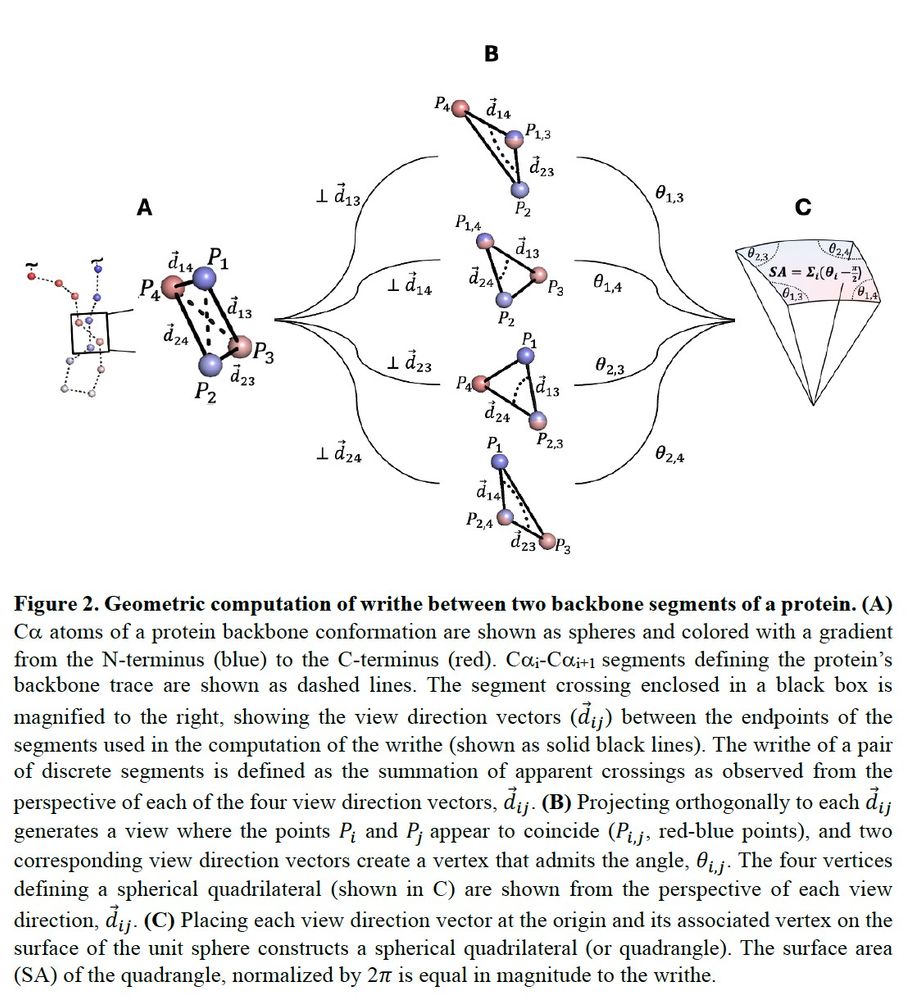
April 30, 2025 at 5:45 PM
Here is an illustration of how to compute writhe in a geometric fashion. You can project the apparent crossings of two segments to define a spherical quadrangle, and use this the accurately calculate writhe. Tommy wrote a python package to do this real fast
github.com/paulrobustel...
github.com/paulrobustel...
You can think of a writhe matrix as having a radial component that describes the distance between segments, with an additional angular component, that describes the relative orientations of each segment. It has the distance information of a contact map, with additional info on segment orientations

April 30, 2025 at 5:45 PM
You can think of a writhe matrix as having a radial component that describes the distance between segments, with an additional angular component, that describes the relative orientations of each segment. It has the distance information of a contact map, with additional info on segment orientations