



Mykhaylo M. Malakhov
@mykmal.xyz
Genetic epidemiology postdoc at @stanford.edu
Me too, especially if the paper has any mathematical notation in it.
November 8, 2025 at 3:05 PM
Me too, especially if the paper has any mathematical notation in it.
More votes have been counted since then, and the updated map looks even better. There's not a single red cone across the whole state!

November 7, 2025 at 9:03 AM
More votes have been counted since then, and the updated map looks even better. There's not a single red cone across the whole state!
Reading papers detracts from time that could be spent actually analyzing data, and yet keeping up with new research is necessary to analyze data effectively. I'm still struggling to find a good balance, especially when I feel like I barely have enough time to read my email...
September 24, 2025 at 6:53 AM
Reading papers detracts from time that could be spent actually analyzing data, and yet keeping up with new research is necessary to analyze data effectively. I'm still struggling to find a good balance, especially when I feel like I barely have enough time to read my email...
Congratulations again on receiving the Williams Award for the best presentation by a student at IGES!! I really enjoyed meeting you and learning about your cool work!
September 3, 2025 at 12:05 PM
Congratulations again on receiving the Williams Award for the best presentation by a student at IGES!! I really enjoyed meeting you and learning about your cool work!
I had exactly the same thought while reading your preprint yesterday! I think that combining our two methods is a very promising idea. I have never initiated a collaboration outside my own lab before, but I would love to pursue this direction together if you are interested!
June 9, 2025 at 7:00 PM
I had exactly the same thought while reading your preprint yesterday! I think that combining our two methods is a very promising idea. I have never initiated a collaboration outside my own lab before, but I would love to pursue this direction together if you are interested!
Very cool work! I'm glad to see others also advocating for moving beyond the marginal, single-gene paradigm. I did a related project for my PhD, extending TWAS to account for genetically regulated co-expression (doi.org/10.1101/2024...). Maybe we can connect once I move to Stanford this fall!

Co-expression-wide association studies link genetically regulated interactions with complex traits
Transcriptome- and proteome-wide association studies (TWAS/PWAS) have proven successful in prioritizing genes and proteins whose genetically regulated expression modulates disease risk, but they ignor...
doi.org
June 8, 2025 at 9:29 PM
Very cool work! I'm glad to see others also advocating for moving beyond the marginal, single-gene paradigm. I did a related project for my PhD, extending TWAS to account for genetically regulated co-expression (doi.org/10.1101/2024...). Maybe we can connect once I move to Stanford this fall!
Skimming papers that use vastly different terminology or notation is not easy either, but I hope you are right that conventions in these fields are starting to converge!
May 15, 2025 at 10:38 PM
Skimming papers that use vastly different terminology or notation is not easy either, but I hope you are right that conventions in these fields are starting to converge!
I am a young person, and I can confirm that I have wondered about this many times. But what is the solution? I've tried searching econometrics journals for papers on IV regression, but I did not get very far because the terminology and notation are completely different from what I'm familiar with.
May 13, 2025 at 11:24 PM
I am a young person, and I can confirm that I have wondered about this many times. But what is the solution? I've tried searching econometrics journals for papers on IV regression, but I did not get very far because the terminology and notation are completely different from what I'm familiar with.
Importantly, type I error was not inflated because imputed data was only used for training, not inference. Overall, we demonstrated that GWAS-based trait imputation can be used to increase sample sizes and thereby boost power for nonlinear TWAS/PWAS methods that require individual-level data. (6/6)
May 7, 2025 at 6:12 PM
Importantly, type I error was not inflated because imputed data was only used for training, not inference. Overall, we demonstrated that GWAS-based trait imputation can be used to increase sample sizes and thereby boost power for nonlinear TWAS/PWAS methods that require individual-level data. (6/6)
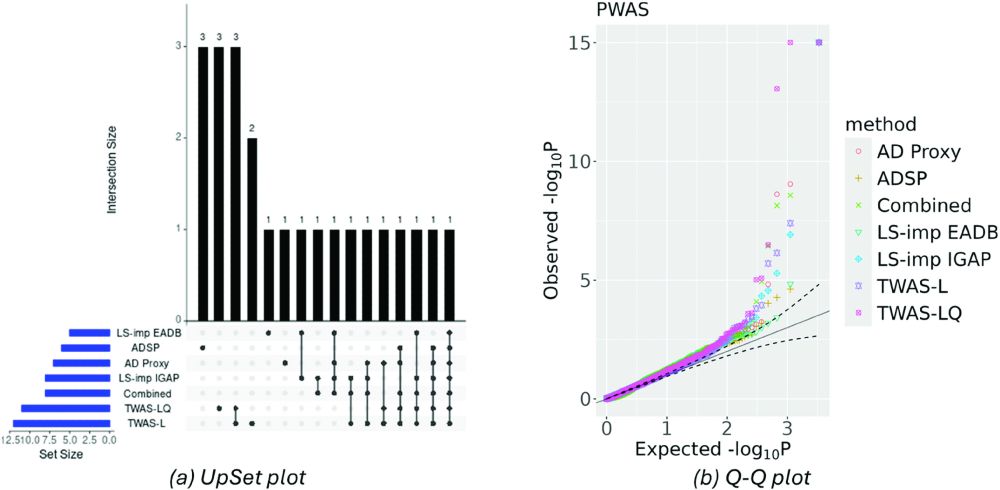
We found that DeLIVR trained on imputed AD status was able to identify disease-relevant genes that were missed if the models were instead trained on observed data, and the same story held true when identifying putative risk proteins. We also showed that LS-imputation has the best performance. (5/6)

May 7, 2025 at 6:12 PM
We found that DeLIVR trained on imputed AD status was able to identify disease-relevant genes that were missed if the models were instead trained on observed data, and the same story held true when identifying putative risk proteins. We also showed that LS-imputation has the best performance. (5/6)
We considered the nonlinear TWAS/PWAS methods DeLIVR (based on deep neural networks) and TWAS-LQ (based on a parametric polynomial model). We focused on Alzheimer's disease (AD) and imputed AD status from family history or from GWAS summary statistics using LS-imputation, PRS-CS, and LDpred2. (4/6)

May 7, 2025 at 6:12 PM
We considered the nonlinear TWAS/PWAS methods DeLIVR (based on deep neural networks) and TWAS-LQ (based on a parametric polynomial model). We focused on Alzheimer's disease (AD) and imputed AD status from family history or from GWAS summary statistics using LS-imputation, PRS-CS, and LDpred2. (4/6)
But linear TWAS/PWAS methods have one major advantage: linear stage 2 models can be estimated using GWAS summary statistics instead of individual-level data. So what if we use GWAS summary statistics to create imputed individual-level datasets and then train nonlinear TWAS/PWAS models on them? (3/6)

May 7, 2025 at 6:12 PM
But linear TWAS/PWAS methods have one major advantage: linear stage 2 models can be estimated using GWAS summary statistics instead of individual-level data. So what if we use GWAS summary statistics to create imputed individual-level datasets and then train nonlinear TWAS/PWAS models on them? (3/6)
Transcriptome- and proteome-wide association studies (TWAS/PWAS) traditionally rely on linear models and can only detect linear effects. Recently our group and others developed IV regression methods based on flexible models, which can identify genes with nonlinear effects on complex traits. (2/6)
May 7, 2025 at 6:12 PM
Transcriptome- and proteome-wide association studies (TWAS/PWAS) traditionally rely on linear models and can only detect linear effects. Recently our group and others developed IV regression methods based on flexible models, which can identify genes with nonlinear effects on complex traits. (2/6)