




Simon Mitchell
@mitchell.science
Cancer systems biology lab @bsmsmedschool.bsky.social | @UKRI.org Future Leaders Fellow | Director of @SussexCancer.org | Frontiers in Immunology Speciality Chief Editor: Systems Immunology
Excited to have been promoted to Professor in Cancer Systems Biology at @bsmsmedschool.bsky.social, University of Sussex, @uniofbrighton.bsky.social
October 9, 2025 at 8:20 PM
Excited to have been promoted to Professor in Cancer Systems Biology at @bsmsmedschool.bsky.social, University of Sussex, @uniofbrighton.bsky.social
Finally, we find that despite all this complexity there is one strategy that always works to overcome the TME.
Inhibition of the key regulator of non-canonical NF-kB (NIK) totally neutralises the drug resistance caused by the TME in DLBCL🎉
5/🧵
Inhibition of the key regulator of non-canonical NF-kB (NIK) totally neutralises the drug resistance caused by the TME in DLBCL🎉
5/🧵

August 18, 2025 at 11:05 AM
Finally, we find that despite all this complexity there is one strategy that always works to overcome the TME.
Inhibition of the key regulator of non-canonical NF-kB (NIK) totally neutralises the drug resistance caused by the TME in DLBCL🎉
5/🧵
Inhibition of the key regulator of non-canonical NF-kB (NIK) totally neutralises the drug resistance caused by the TME in DLBCL🎉
5/🧵
We used computational models, ChIP-seq, imaging, phospho-protemics, and mouse models (collab with Alexander Hoffmann) to discover that high BCR signalling in these cells re-wires the NF-kB:BCL2 network.
Good news: this means Ibrutinib overcomes the TME!
4/🧵
Good news: this means Ibrutinib overcomes the TME!
4/🧵

August 18, 2025 at 11:05 AM
We used computational models, ChIP-seq, imaging, phospho-protemics, and mouse models (collab with Alexander Hoffmann) to discover that high BCR signalling in these cells re-wires the NF-kB:BCL2 network.
Good news: this means Ibrutinib overcomes the TME!
4/🧵
Good news: this means Ibrutinib overcomes the TME!
4/🧵
We find:
- culturing DLBCL cells with cells expressing CD40 (to mimick the TME) creates resistance to BH3-mimetics.
- this happens whether you're targeting BCL2 or BCLXL.
- Compensatory upregulation of BCLXL causes resistance.
- In SUDHL8 cells MCL1 also increases. Why!?
3/🧵
- culturing DLBCL cells with cells expressing CD40 (to mimick the TME) creates resistance to BH3-mimetics.
- this happens whether you're targeting BCL2 or BCLXL.
- Compensatory upregulation of BCLXL causes resistance.
- In SUDHL8 cells MCL1 also increases. Why!?
3/🧵

August 18, 2025 at 11:05 AM
We find:
- culturing DLBCL cells with cells expressing CD40 (to mimick the TME) creates resistance to BH3-mimetics.
- this happens whether you're targeting BCL2 or BCLXL.
- Compensatory upregulation of BCLXL causes resistance.
- In SUDHL8 cells MCL1 also increases. Why!?
3/🧵
- culturing DLBCL cells with cells expressing CD40 (to mimick the TME) creates resistance to BH3-mimetics.
- this happens whether you're targeting BCL2 or BCLXL.
- Compensatory upregulation of BCLXL causes resistance.
- In SUDHL8 cells MCL1 also increases. Why!?
3/🧵
Background:
- Established NFκB -> BCL2 link is actually a bit fuzzy (which NFκB subunits control which BCL2 proteins in DLBCL?)
- scRNAseq suggests tumour microenvironment (TME) activates non-canonical NFκB (RelB:p52).
- Can we overcome the protective effect of the TME ?
2/🧵
- Established NFκB -> BCL2 link is actually a bit fuzzy (which NFκB subunits control which BCL2 proteins in DLBCL?)
- scRNAseq suggests tumour microenvironment (TME) activates non-canonical NFκB (RelB:p52).
- Can we overcome the protective effect of the TME ?
2/🧵

August 18, 2025 at 11:05 AM
Background:
- Established NFκB -> BCL2 link is actually a bit fuzzy (which NFκB subunits control which BCL2 proteins in DLBCL?)
- scRNAseq suggests tumour microenvironment (TME) activates non-canonical NFκB (RelB:p52).
- Can we overcome the protective effect of the TME ?
2/🧵
- Established NFκB -> BCL2 link is actually a bit fuzzy (which NFκB subunits control which BCL2 proteins in DLBCL?)
- scRNAseq suggests tumour microenvironment (TME) activates non-canonical NFκB (RelB:p52).
- Can we overcome the protective effect of the TME ?
2/🧵
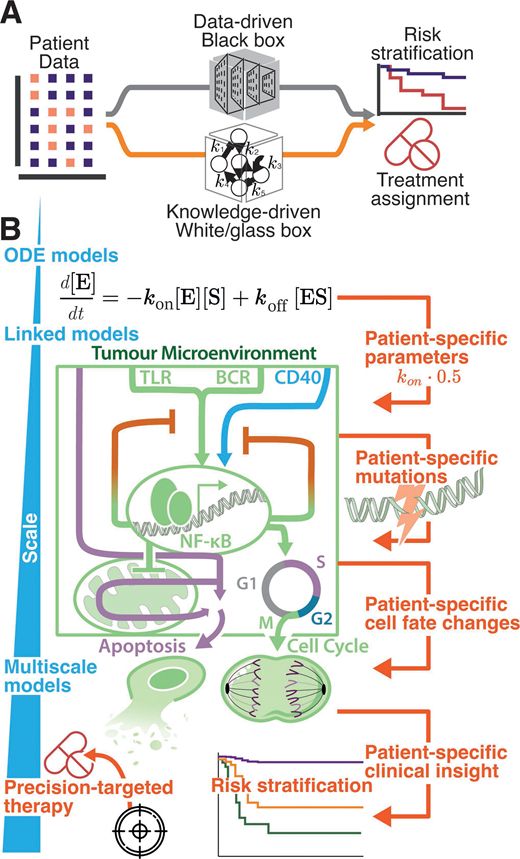
New paper led by @ejayawant.bsky.social 🎉
We review:
- how computational biology is helping us understand lymphoma,
- how machine learning and cell simulations make sense of vast amounts of data,
- how models can personalise treatments to each person with lymphoma.
portlandpress.com/biochemsoctr...
We review:
- how computational biology is helping us understand lymphoma,
- how machine learning and cell simulations make sense of vast amounts of data,
- how models can personalise treatments to each person with lymphoma.
portlandpress.com/biochemsoctr...
July 4, 2025 at 4:02 PM
New paper led by @ejayawant.bsky.social 🎉
We review:
- how computational biology is helping us understand lymphoma,
- how machine learning and cell simulations make sense of vast amounts of data,
- how models can personalise treatments to each person with lymphoma.
portlandpress.com/biochemsoctr...
We review:
- how computational biology is helping us understand lymphoma,
- how machine learning and cell simulations make sense of vast amounts of data,
- how models can personalise treatments to each person with lymphoma.
portlandpress.com/biochemsoctr...
so lovely to be at @cshlnews.bsky.social for the Systems Immunology Meeting.
Great venue and even better science.
Great venue and even better science.

April 23, 2025 at 7:08 PM
so lovely to be at @cshlnews.bsky.social for the Systems Immunology Meeting.
Great venue and even better science.
Great venue and even better science.
Proud moment from Arran Pack's graduation.
It's so rewarding to see our lab's first PhD student graduate, and I couldn't have asked for a better first student.
Job well done Arran!
🤜🤛
It's so rewarding to see our lab's first PhD student graduate, and I couldn't have asked for a better first student.
Job well done Arran!
🤜🤛

February 14, 2025 at 12:10 PM
Proud moment from Arran Pack's graduation.
It's so rewarding to see our lab's first PhD student graduate, and I couldn't have asked for a better first student.
Job well done Arran!
🤜🤛
It's so rewarding to see our lab's first PhD student graduate, and I couldn't have asked for a better first student.
Job well done Arran!
🤜🤛
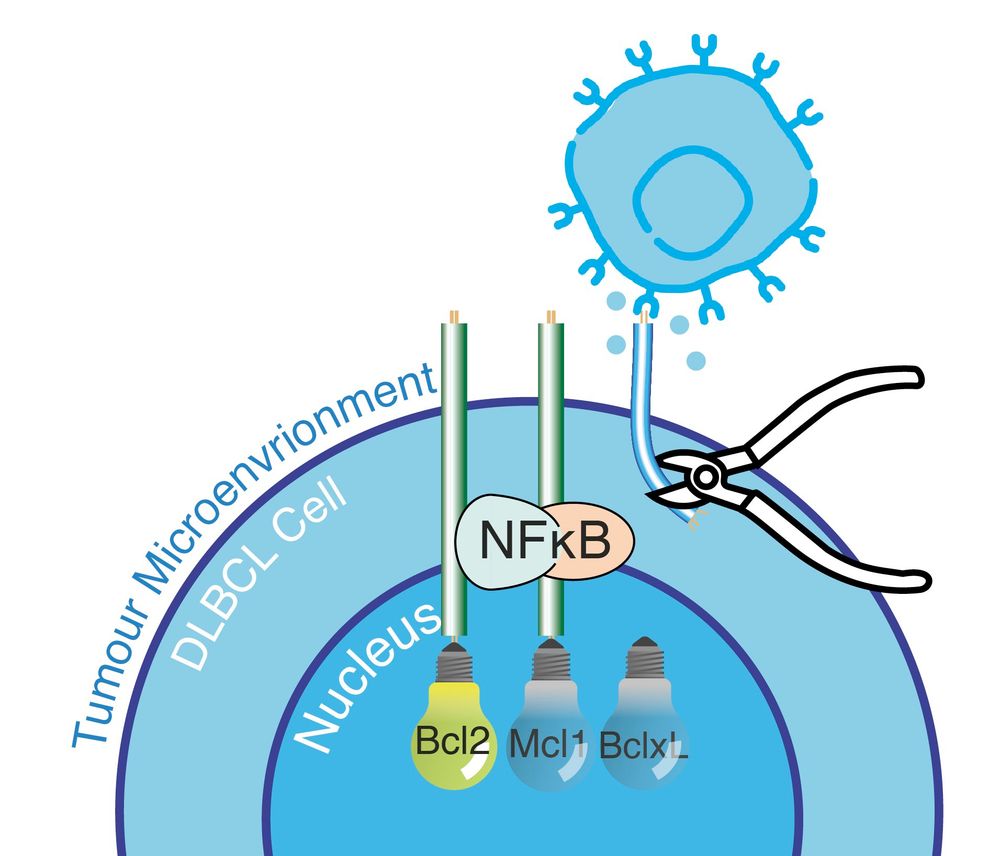
Yes!
Non-canonical NF-kB inhibition with Amgen16 overcomes all the microenvrionment mediated resistance we discovered, regardless of the mechanism!
@amgen.bsky.social
Let's summarise...
Non-canonical NF-kB inhibition with Amgen16 overcomes all the microenvrionment mediated resistance we discovered, regardless of the mechanism!
@amgen.bsky.social
Let's summarise...
December 6, 2024 at 6:18 PM
Yes!
Non-canonical NF-kB inhibition with Amgen16 overcomes all the microenvrionment mediated resistance we discovered, regardless of the mechanism!
@amgen.bsky.social
Let's summarise...
Non-canonical NF-kB inhibition with Amgen16 overcomes all the microenvrionment mediated resistance we discovered, regardless of the mechanism!
@amgen.bsky.social
Let's summarise...
So, taking a step back, we have:
- Different Achilles heel in each DLBCL
- Different resistance mechanism mediated by complicated cross talk.
This makes it really hard to improve treatments.
We asked if we could break all the links with the TME by inhibiting non-canonical NF-kB?
- Different Achilles heel in each DLBCL
- Different resistance mechanism mediated by complicated cross talk.
This makes it really hard to improve treatments.
We asked if we could break all the links with the TME by inhibiting non-canonical NF-kB?
December 6, 2024 at 6:18 PM
So, taking a step back, we have:
- Different Achilles heel in each DLBCL
- Different resistance mechanism mediated by complicated cross talk.
This makes it really hard to improve treatments.
We asked if we could break all the links with the TME by inhibiting non-canonical NF-kB?
- Different Achilles heel in each DLBCL
- Different resistance mechanism mediated by complicated cross talk.
This makes it really hard to improve treatments.
We asked if we could break all the links with the TME by inhibiting non-canonical NF-kB?
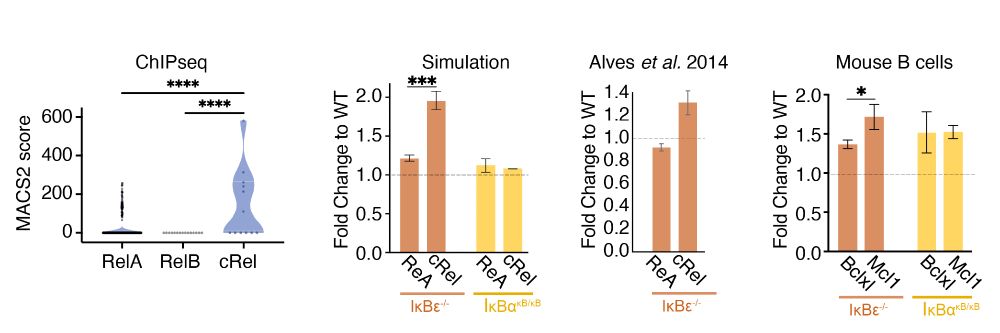
ChIPseq says yes, but we needed another test.
The model told us an IkBe knockout would selectively upregulate cRel, which would upregulate MCL1 if we were right.
Collaborating with Alexander Hoffmann's group we tested this in IkBe KO mice.
cRel and MCL1 go up!
The model told us an IkBe knockout would selectively upregulate cRel, which would upregulate MCL1 if we were right.
Collaborating with Alexander Hoffmann's group we tested this in IkBe KO mice.
cRel and MCL1 go up!
December 6, 2024 at 6:18 PM
ChIPseq says yes, but we needed another test.
The model told us an IkBe knockout would selectively upregulate cRel, which would upregulate MCL1 if we were right.
Collaborating with Alexander Hoffmann's group we tested this in IkBe KO mice.
cRel and MCL1 go up!
The model told us an IkBe knockout would selectively upregulate cRel, which would upregulate MCL1 if we were right.
Collaborating with Alexander Hoffmann's group we tested this in IkBe KO mice.
cRel and MCL1 go up!
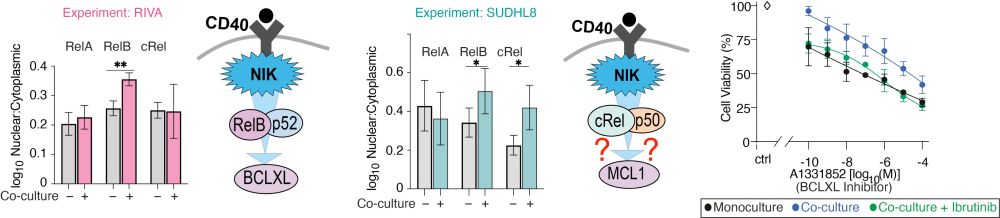
Imaging confirms it!
Chronic NF-kB activation creates crosstalk between the pathways.
We also knew this chronic NF-kB was causing the drug resistance, because when we inhibit it (with ibrutinib) we can re-sensitize the cells.
But the link between cRel and MCL1 is new.
Is it real?
Chronic NF-kB activation creates crosstalk between the pathways.
We also knew this chronic NF-kB was causing the drug resistance, because when we inhibit it (with ibrutinib) we can re-sensitize the cells.
But the link between cRel and MCL1 is new.
Is it real?
December 6, 2024 at 6:18 PM
Imaging confirms it!
Chronic NF-kB activation creates crosstalk between the pathways.
We also knew this chronic NF-kB was causing the drug resistance, because when we inhibit it (with ibrutinib) we can re-sensitize the cells.
But the link between cRel and MCL1 is new.
Is it real?
Chronic NF-kB activation creates crosstalk between the pathways.
We also knew this chronic NF-kB was causing the drug resistance, because when we inhibit it (with ibrutinib) we can re-sensitize the cells.
But the link between cRel and MCL1 is new.
Is it real?
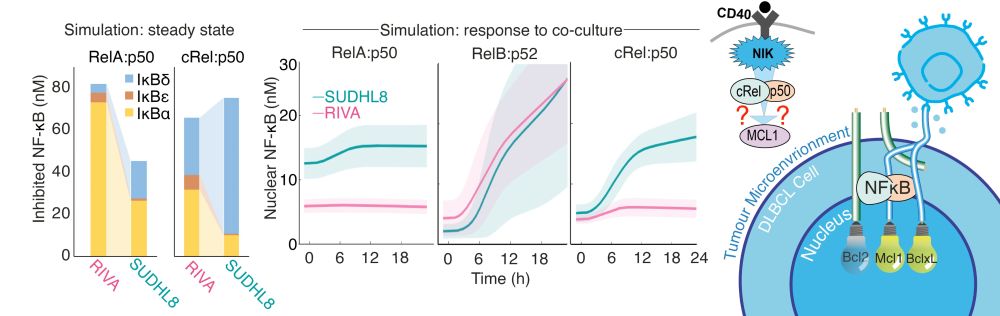
Enter computational modelling.
A model developed by Arran Pack
predicted that the high RelA could create cross talk between the NF-kB pathways.
CD40 -> NIK -> cRel -> MCL1?
Soumen Basak has seen this before in healthy B-cells. We predict it's happening in DLBCL.
Let's check
A model developed by Arran Pack
predicted that the high RelA could create cross talk between the NF-kB pathways.
CD40 -> NIK -> cRel -> MCL1?
Soumen Basak has seen this before in healthy B-cells. We predict it's happening in DLBCL.
Let's check
December 6, 2024 at 6:18 PM
Enter computational modelling.
A model developed by Arran Pack
predicted that the high RelA could create cross talk between the NF-kB pathways.
CD40 -> NIK -> cRel -> MCL1?
Soumen Basak has seen this before in healthy B-cells. We predict it's happening in DLBCL.
Let's check
A model developed by Arran Pack
predicted that the high RelA could create cross talk between the NF-kB pathways.
CD40 -> NIK -> cRel -> MCL1?
Soumen Basak has seen this before in healthy B-cells. We predict it's happening in DLBCL.
Let's check
We knew the MCL1 was probably NF-kB's fault, isn't everything ;)
We used imaging to find which NF-kB components were in the nucleus before the cells went into co-culture.
Lots of RelA in cells that induce MCL1.
But how does high RelA in monoculture lead to MCL1 in co-culture?
We used imaging to find which NF-kB components were in the nucleus before the cells went into co-culture.
Lots of RelA in cells that induce MCL1.
But how does high RelA in monoculture lead to MCL1 in co-culture?

December 6, 2024 at 6:18 PM
We knew the MCL1 was probably NF-kB's fault, isn't everything ;)
We used imaging to find which NF-kB components were in the nucleus before the cells went into co-culture.
Lots of RelA in cells that induce MCL1.
But how does high RelA in monoculture lead to MCL1 in co-culture?
We used imaging to find which NF-kB components were in the nucleus before the cells went into co-culture.
Lots of RelA in cells that induce MCL1.
But how does high RelA in monoculture lead to MCL1 in co-culture?
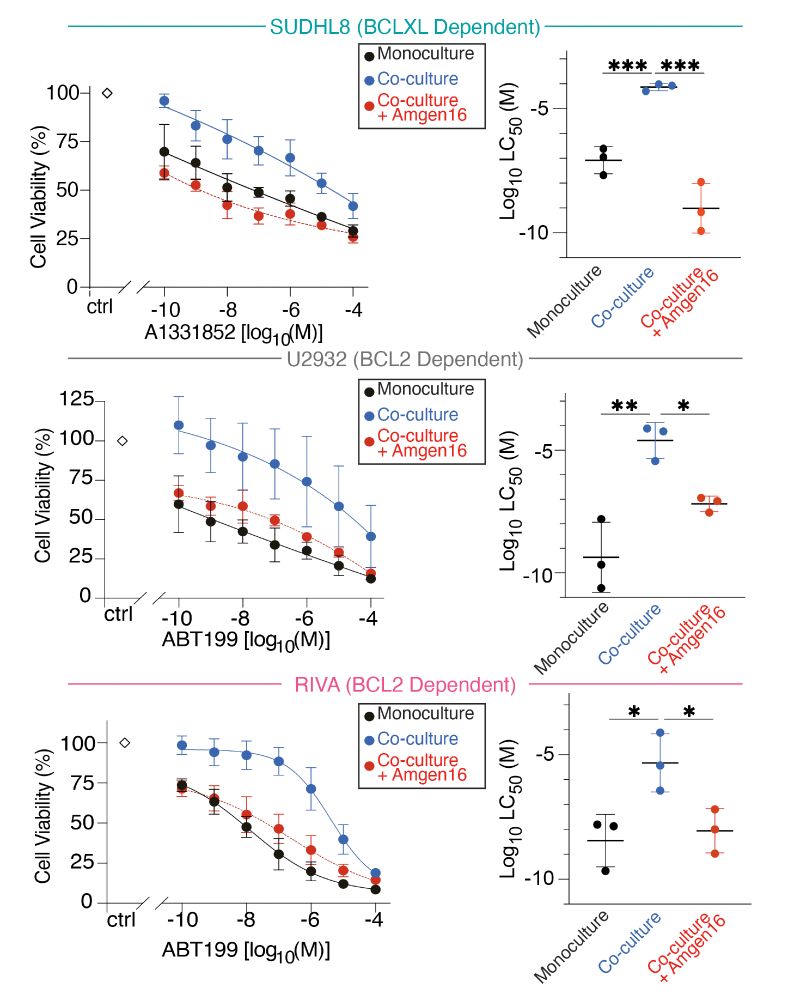
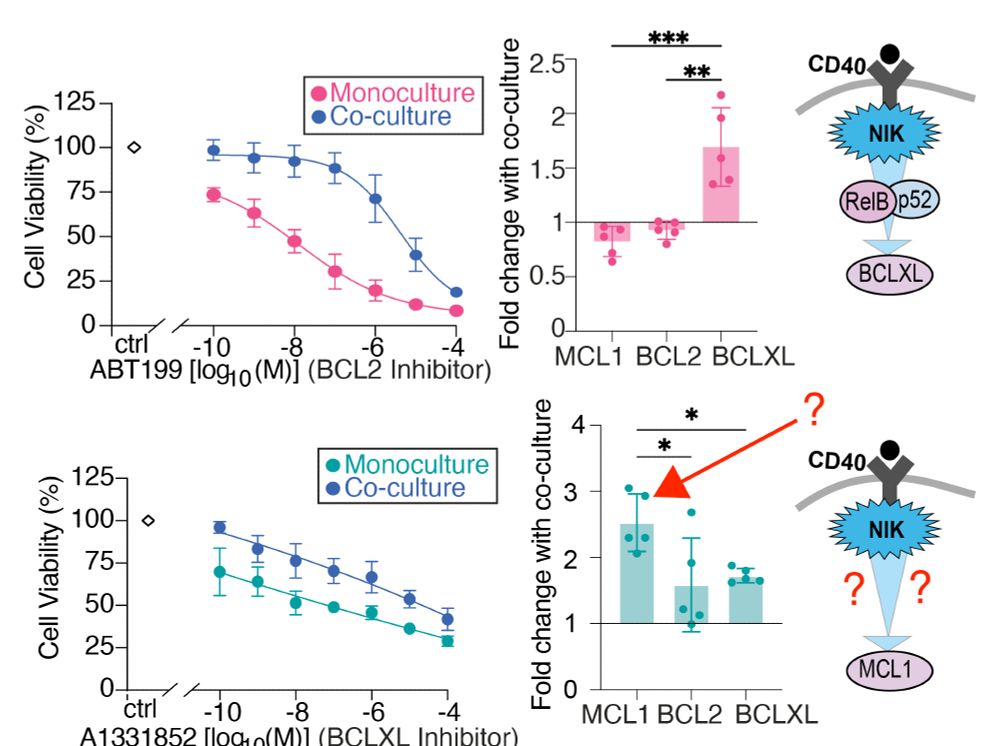
The co-culture effect was seen in multiple cell lines, to drugs targetting BCL2 and BCLXL. This might explain why these drugs work in the lab but not in DLBCL patients.
Some of this we understood:
CD40 -> NIK -> RelB -> BCLXL
But some really did not:
CD40 -> NIK -> ? -> MCL!?
Some of this we understood:
CD40 -> NIK -> RelB -> BCLXL
But some really did not:
CD40 -> NIK -> ? -> MCL!?
December 6, 2024 at 6:18 PM
The co-culture effect was seen in multiple cell lines, to drugs targetting BCL2 and BCLXL. This might explain why these drugs work in the lab but not in DLBCL patients.
Some of this we understood:
CD40 -> NIK -> RelB -> BCLXL
But some really did not:
CD40 -> NIK -> ? -> MCL!?
Some of this we understood:
CD40 -> NIK -> RelB -> BCLXL
But some really did not:
CD40 -> NIK -> ? -> MCL!?
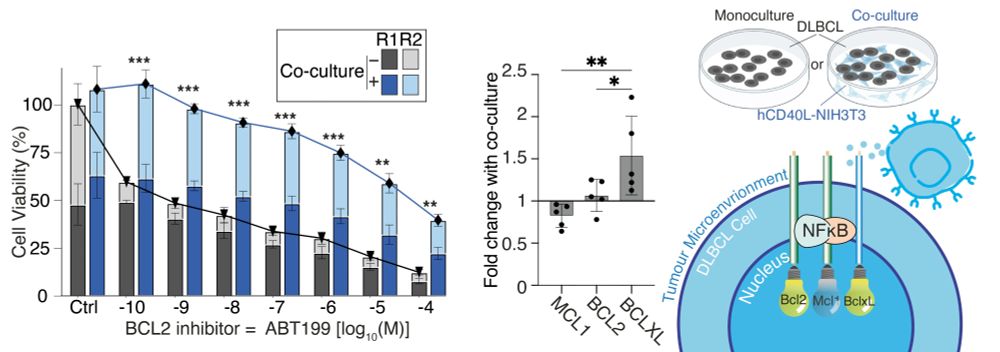
We used a co-culture system that mimicks how T-cells support DLBCL cells in lymph nodes.
This made both cell populations resist treatment. They upregulated BCLXL which compensates for BCL2 inhibition.
Inherent resistance, and micro-environmental resistance in one cell line.
Neat
This made both cell populations resist treatment. They upregulated BCLXL which compensates for BCL2 inhibition.
Inherent resistance, and micro-environmental resistance in one cell line.
Neat
December 6, 2024 at 6:18 PM
We used a co-culture system that mimicks how T-cells support DLBCL cells in lymph nodes.
This made both cell populations resist treatment. They upregulated BCLXL which compensates for BCL2 inhibition.
Inherent resistance, and micro-environmental resistance in one cell line.
Neat
This made both cell populations resist treatment. They upregulated BCLXL which compensates for BCL2 inhibition.
Inherent resistance, and micro-environmental resistance in one cell line.
Neat
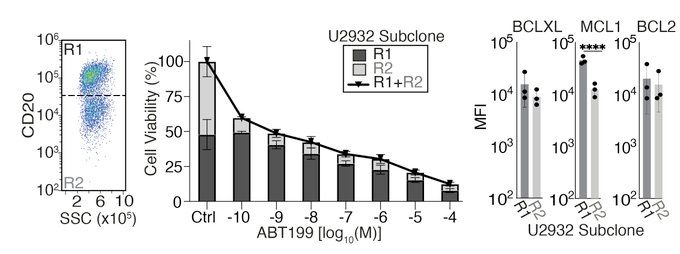
We find the two cell populations in the same cell line have totally different inherent sensitivity to BCL2 inhibition!
Why?
One has high MCL1 to compensate for BCL2 inhibition.
This is cool, but these are cells in a dish. Could we still drug these in a supportive lymph node?
Why?
One has high MCL1 to compensate for BCL2 inhibition.
This is cool, but these are cells in a dish. Could we still drug these in a supportive lymph node?
December 6, 2024 at 6:18 PM
We find the two cell populations in the same cell line have totally different inherent sensitivity to BCL2 inhibition!
Why?
One has high MCL1 to compensate for BCL2 inhibition.
This is cool, but these are cells in a dish. Could we still drug these in a supportive lymph node?
Why?
One has high MCL1 to compensate for BCL2 inhibition.
This is cool, but these are cells in a dish. Could we still drug these in a supportive lymph node?
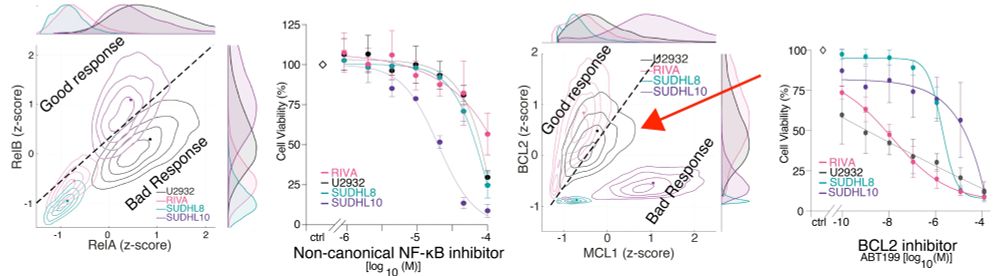
Do these fingerprints predict drug responses?
Yes!
But what is going on with the U2932 cell line? Like lots of cancer it contains multiple cell populations with different genetics.
This makes it great for figuring out how different cells in the same tumour might respond to a drug.
Yes!
But what is going on with the U2932 cell line? Like lots of cancer it contains multiple cell populations with different genetics.
This makes it great for figuring out how different cells in the same tumour might respond to a drug.
December 6, 2024 at 6:18 PM
Do these fingerprints predict drug responses?
Yes!
But what is going on with the U2932 cell line? Like lots of cancer it contains multiple cell populations with different genetics.
This makes it great for figuring out how different cells in the same tumour might respond to a drug.
Yes!
But what is going on with the U2932 cell line? Like lots of cancer it contains multiple cell populations with different genetics.
This makes it great for figuring out how different cells in the same tumour might respond to a drug.
Diffuse Large B-cell Lymphoma (DLBCL) has lots of BCL2 and NF-kB. Both drive poor prognosis.
Lots of people have tried inhibiting these. But NF-kB is 2 pathways, 5 proteins, and there are many BCL2-family proteins.
We found previously (doi.org/10.3389/fonc...), each DLBCL has a unique "fingerprint".
Lots of people have tried inhibiting these. But NF-kB is 2 pathways, 5 proteins, and there are many BCL2-family proteins.
We found previously (doi.org/10.3389/fonc...), each DLBCL has a unique "fingerprint".

December 6, 2024 at 6:18 PM
Diffuse Large B-cell Lymphoma (DLBCL) has lots of BCL2 and NF-kB. Both drive poor prognosis.
Lots of people have tried inhibiting these. But NF-kB is 2 pathways, 5 proteins, and there are many BCL2-family proteins.
We found previously (doi.org/10.3389/fonc...), each DLBCL has a unique "fingerprint".
Lots of people have tried inhibiting these. But NF-kB is 2 pathways, 5 proteins, and there are many BCL2-family proteins.
We found previously (doi.org/10.3389/fonc...), each DLBCL has a unique "fingerprint".
Excited to share a new pre-print led by the incredible PhD student Aimilia Vareli.
biorxiv.org/content/10.1...
She asked why drugs that kill cancer cells in blood don't kill them in lymph nodes? How we can fix that?
🧵🧪
@sussexcancer.org
@bsmsmedschool.bsky.social @sussexuni.bsky.social
biorxiv.org/content/10.1...
She asked why drugs that kill cancer cells in blood don't kill them in lymph nodes? How we can fix that?
🧵🧪
@sussexcancer.org
@bsmsmedschool.bsky.social @sussexuni.bsky.social

December 6, 2024 at 6:18 PM
Excited to share a new pre-print led by the incredible PhD student Aimilia Vareli.
biorxiv.org/content/10.1...
She asked why drugs that kill cancer cells in blood don't kill them in lymph nodes? How we can fix that?
🧵🧪
@sussexcancer.org
@bsmsmedschool.bsky.social @sussexuni.bsky.social
biorxiv.org/content/10.1...
She asked why drugs that kill cancer cells in blood don't kill them in lymph nodes? How we can fix that?
🧵🧪
@sussexcancer.org
@bsmsmedschool.bsky.social @sussexuni.bsky.social