



Kevin K. Yang 楊凱筌
@kevinkaichuang.bsky.social
Principal Researcher in BioML at Microsoft Research. He/him/他. 🇹🇼 yangkky.github.io
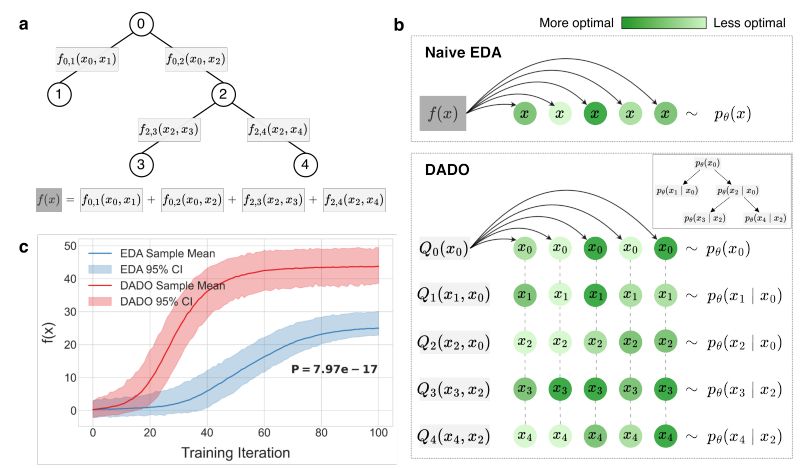
Many sequence-fitness functions are factorizable in sequence space. Taking advantage of this enables more efficient optimization by shrinking the effective search space.
James C Bowden, Sergey Levine @jlistgarten.bsky.social
arxiv.org/abs/2511.03032
James C Bowden, Sergey Levine @jlistgarten.bsky.social
arxiv.org/abs/2511.03032

November 11, 2025 at 3:34 PM
Many sequence-fitness functions are factorizable in sequence space. Taking advantage of this enables more efficient optimization by shrinking the effective search space.
James C Bowden, Sergey Levine @jlistgarten.bsky.social
arxiv.org/abs/2511.03032
James C Bowden, Sergey Levine @jlistgarten.bsky.social
arxiv.org/abs/2511.03032
Design stable, folded proteins using only the 10 "ancient" amino acids.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...


October 31, 2025 at 7:40 PM
Design stable, folded proteins using only the 10 "ancient" amino acids.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
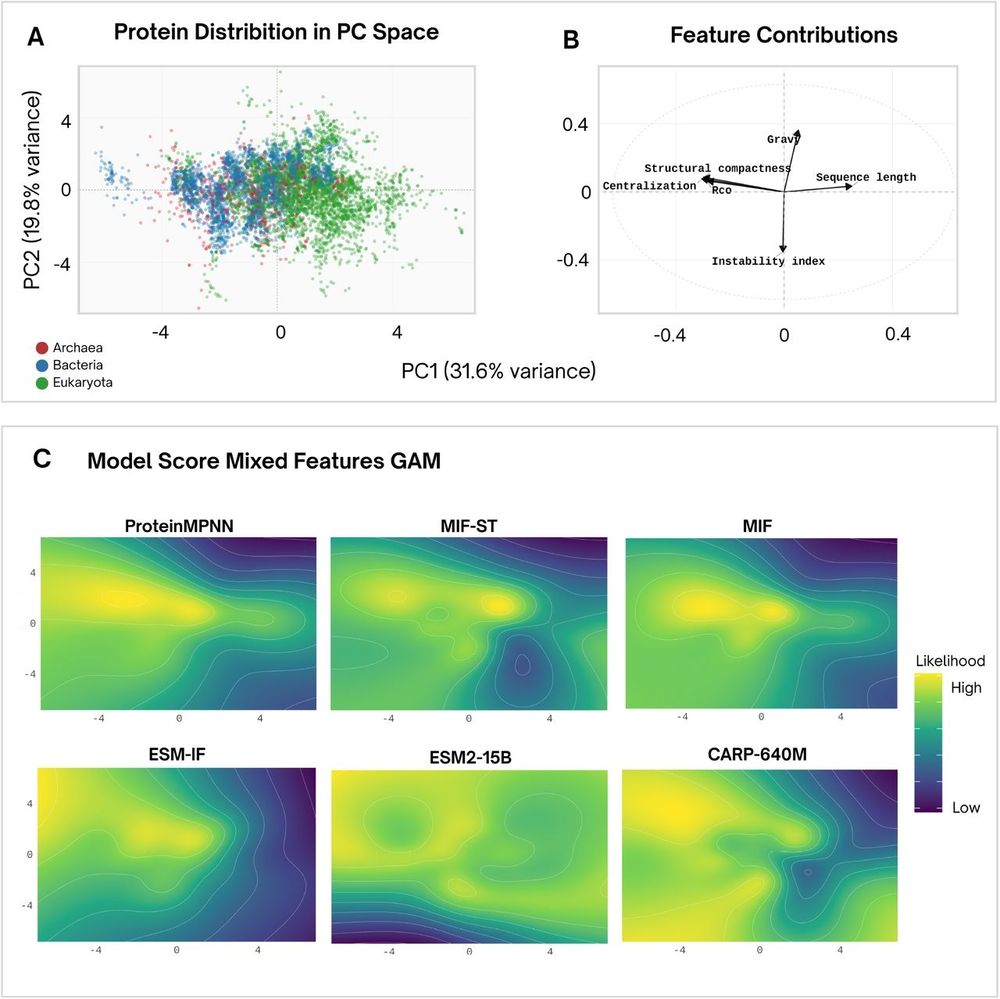
Structure - > sequence models have different phylogenetic (and biochemical) preferences than protein language models.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...



October 29, 2025 at 9:59 PM
Structure - > sequence models have different phylogenetic (and biochemical) preferences than protein language models.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
Alternate between predicting structure with AF3 and designing sequence with ProteinMPNN to generate proteins, including protein, small molecule and nucleic acid binders
@yehlincho.bsky.social @sokrypton.org
www.biorxiv.org/content/10.1...
@yehlincho.bsky.social @sokrypton.org
www.biorxiv.org/content/10.1...



October 15, 2025 at 10:01 PM
Alternate between predicting structure with AF3 and designing sequence with ProteinMPNN to generate proteins, including protein, small molecule and nucleic acid binders
@yehlincho.bsky.social @sokrypton.org
www.biorxiv.org/content/10.1...
@yehlincho.bsky.social @sokrypton.org
www.biorxiv.org/content/10.1...
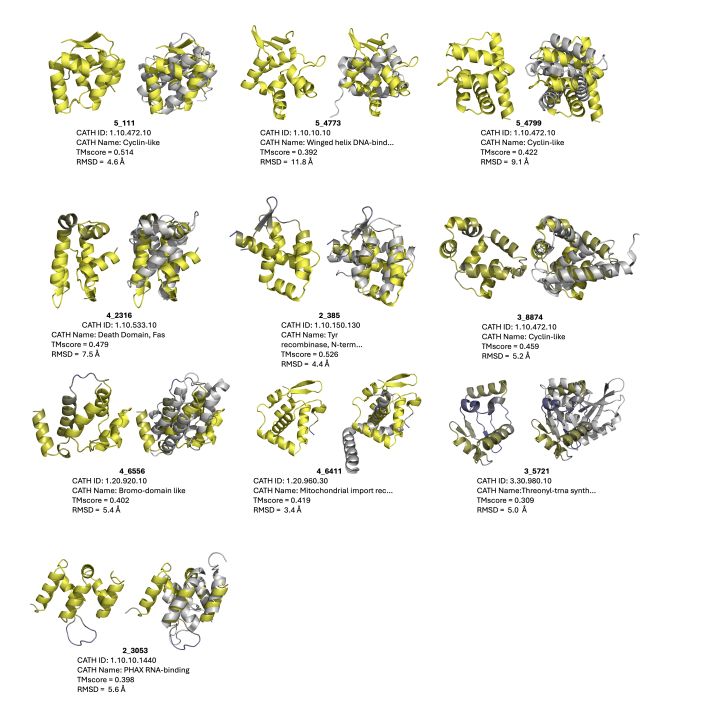
Protein language models can be finetuned to generate many novel structural folds
Arjuna Subramanian, Matt Thomson
www.biorxiv.org/content/10.1...
Arjuna Subramanian, Matt Thomson
www.biorxiv.org/content/10.1...


October 13, 2025 at 2:10 PM
Protein language models can be finetuned to generate many novel structural folds
Arjuna Subramanian, Matt Thomson
www.biorxiv.org/content/10.1...
Arjuna Subramanian, Matt Thomson
www.biorxiv.org/content/10.1...
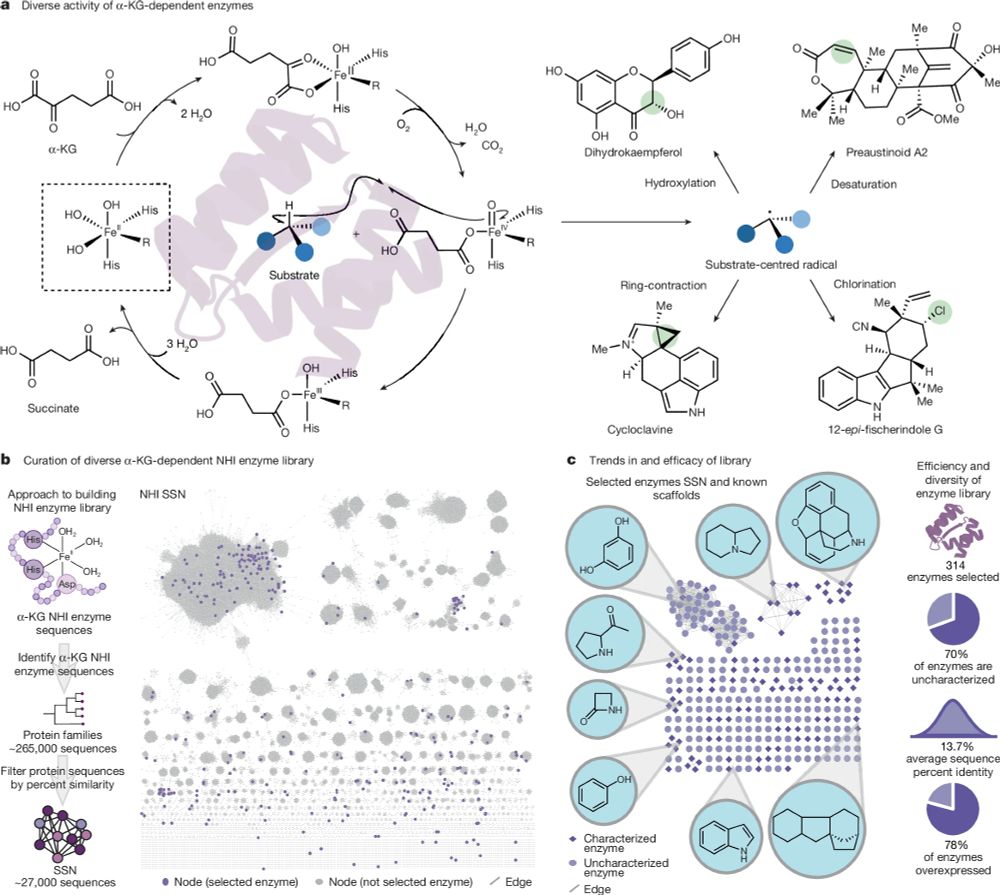
Test the activity of 300+ natural enzymes against 100+ substrates, discover 200+ new enzymatic reactions, and train machine learning models to predict which enzymes can do which reactions.
@aepaton.bsky.social @gabegomes.bsky.social @alisonnarayan.bsky.social
www.nature.com/articles/s41...
@aepaton.bsky.social @gabegomes.bsky.social @alisonnarayan.bsky.social
www.nature.com/articles/s41...

October 2, 2025 at 8:11 PM
Test the activity of 300+ natural enzymes against 100+ substrates, discover 200+ new enzymatic reactions, and train machine learning models to predict which enzymes can do which reactions.
@aepaton.bsky.social @gabegomes.bsky.social @alisonnarayan.bsky.social
www.nature.com/articles/s41...
@aepaton.bsky.social @gabegomes.bsky.social @alisonnarayan.bsky.social
www.nature.com/articles/s41...
Combine multimer structure prediction and an antibody language model to design de novo antibodies with nanomolar binding affinity.
@synbiogaolab.bsky.social @brianhie.bsky.social
www.biorxiv.org/content/10.1...
@synbiogaolab.bsky.social @brianhie.bsky.social
www.biorxiv.org/content/10.1...



September 25, 2025 at 8:02 PM
Combine multimer structure prediction and an antibody language model to design de novo antibodies with nanomolar binding affinity.
@synbiogaolab.bsky.social @brianhie.bsky.social
www.biorxiv.org/content/10.1...
@synbiogaolab.bsky.social @brianhie.bsky.social
www.biorxiv.org/content/10.1...
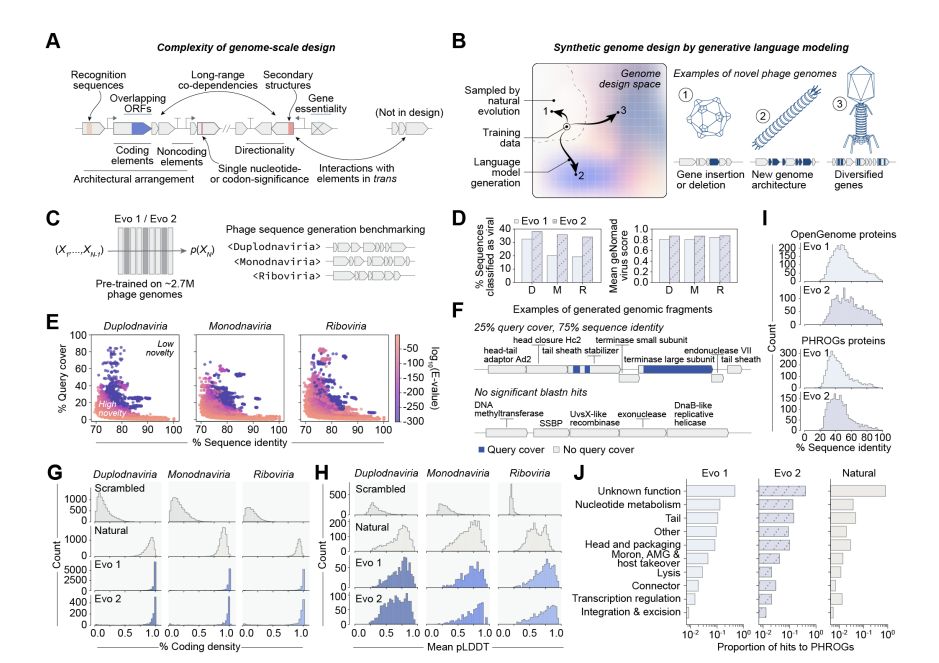
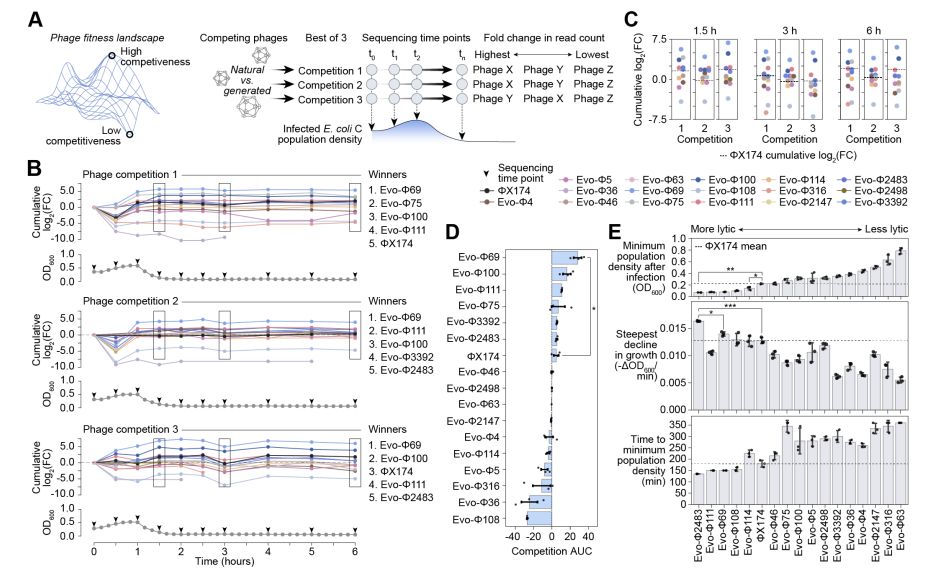
Genome language models can generate new, high-fitness bacteriophages!
@samuelhking.bsky.social @claudiadriscoll.bsky.social
@david-li.bsky.social @danguo.bsky.social @adititm.bsky.social Garyk Brixi @maxewilkinson.bsky.social @brianhie.bsky.social
www.biorxiv.org/content/10.1...
@samuelhking.bsky.social @claudiadriscoll.bsky.social
@david-li.bsky.social @danguo.bsky.social @adititm.bsky.social Garyk Brixi @maxewilkinson.bsky.social @brianhie.bsky.social
www.biorxiv.org/content/10.1...


September 17, 2025 at 9:15 PM
Genome language models can generate new, high-fitness bacteriophages!
@samuelhking.bsky.social @claudiadriscoll.bsky.social
@david-li.bsky.social @danguo.bsky.social @adititm.bsky.social Garyk Brixi @maxewilkinson.bsky.social @brianhie.bsky.social
www.biorxiv.org/content/10.1...
@samuelhking.bsky.social @claudiadriscoll.bsky.social
@david-li.bsky.social @danguo.bsky.social @adititm.bsky.social Garyk Brixi @maxewilkinson.bsky.social @brianhie.bsky.social
www.biorxiv.org/content/10.1...
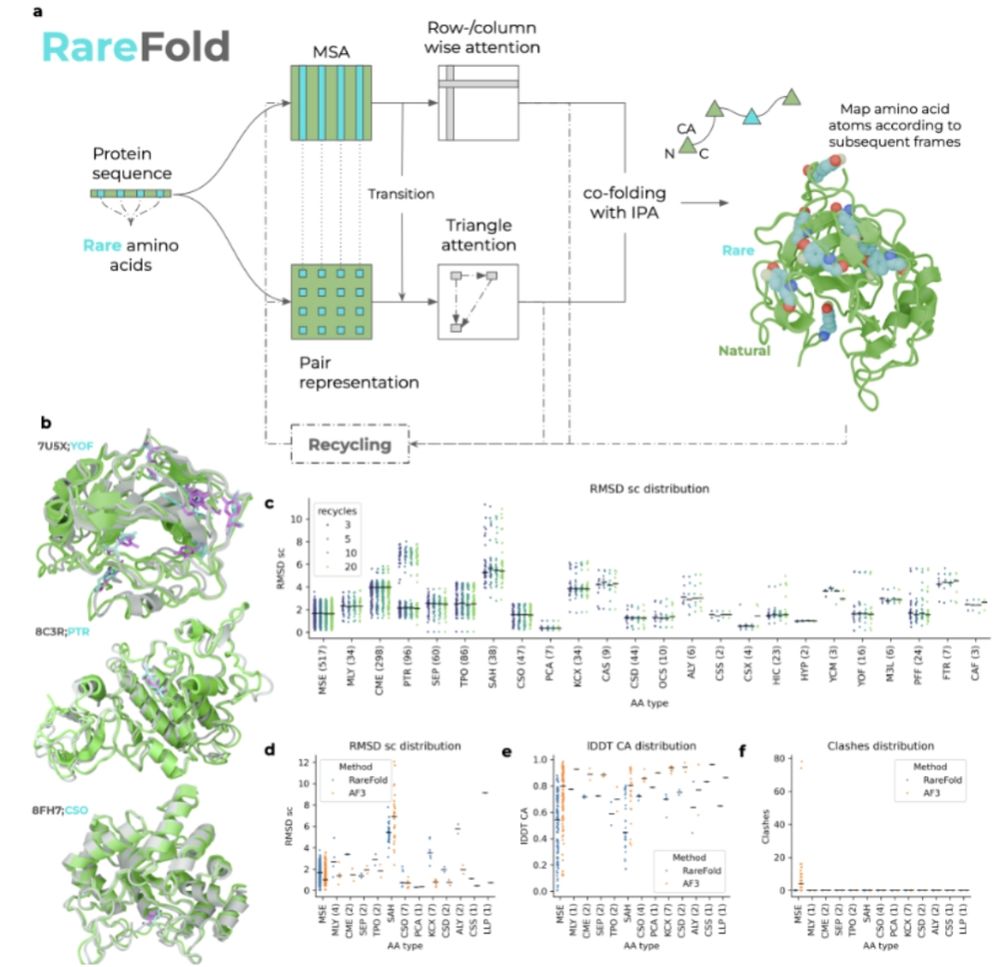
Train a protein structure predictor that can handle 29 non-canonical amino acids, then use it to design binders with non-canonical amino acids that reduce immunogenicity.
@panhammarstrom.bsky.social @patrickbryant1.bsky.social
www.biorxiv.org/content/10.1...
@panhammarstrom.bsky.social @patrickbryant1.bsky.social
www.biorxiv.org/content/10.1...

September 9, 2025 at 8:03 PM
Train a protein structure predictor that can handle 29 non-canonical amino acids, then use it to design binders with non-canonical amino acids that reduce immunogenicity.
@panhammarstrom.bsky.social @patrickbryant1.bsky.social
www.biorxiv.org/content/10.1...
@panhammarstrom.bsky.social @patrickbryant1.bsky.social
www.biorxiv.org/content/10.1...
A joint sequence-structure diffusion model for transmembrane proteins!
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...


September 5, 2025 at 8:18 PM
A joint sequence-structure diffusion model for transmembrane proteins!
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
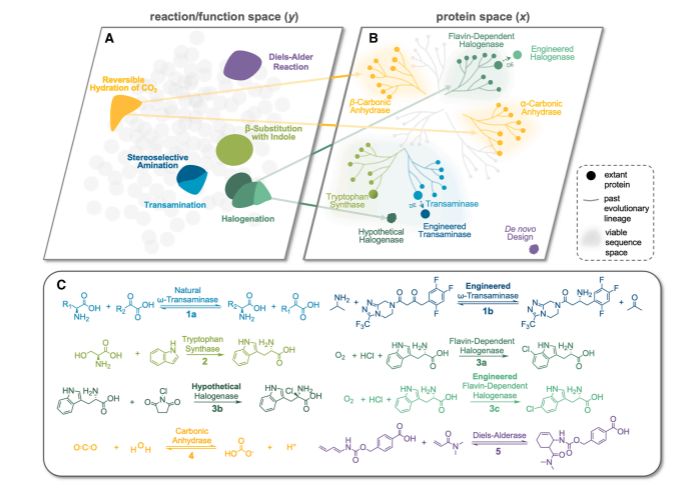
A compelling review of how ML/AI could help in the quest to find an enzyme for every reaction.
@jsunn-y.bsky.social @francescazfl.bsky.social Yueming Long @francesarnold.bsky.social
www.cell.com/cell-systems...
@jsunn-y.bsky.social @francescazfl.bsky.social Yueming Long @francesarnold.bsky.social
www.cell.com/cell-systems...

September 3, 2025 at 12:47 PM
A compelling review of how ML/AI could help in the quest to find an enzyme for every reaction.
@jsunn-y.bsky.social @francescazfl.bsky.social Yueming Long @francesarnold.bsky.social
www.cell.com/cell-systems...
@jsunn-y.bsky.social @francescazfl.bsky.social Yueming Long @francesarnold.bsky.social
www.cell.com/cell-systems...
Typical Ohio activity

August 25, 2025 at 12:12 PM
Typical Ohio activity
The @uwproteindesign.bsky.social's experimental pipeline behind models like RFdiffusion and ProteinMPNN:
- A rapid, scalable, pipeline for producing and characterizing proteins
- A demultiplexing protocol for converting oligopools to clonal constructs
Jason Qian @lfmilles.bsky.social Basile Wicky
- A rapid, scalable, pipeline for producing and characterizing proteins
- A demultiplexing protocol for converting oligopools to clonal constructs
Jason Qian @lfmilles.bsky.social Basile Wicky
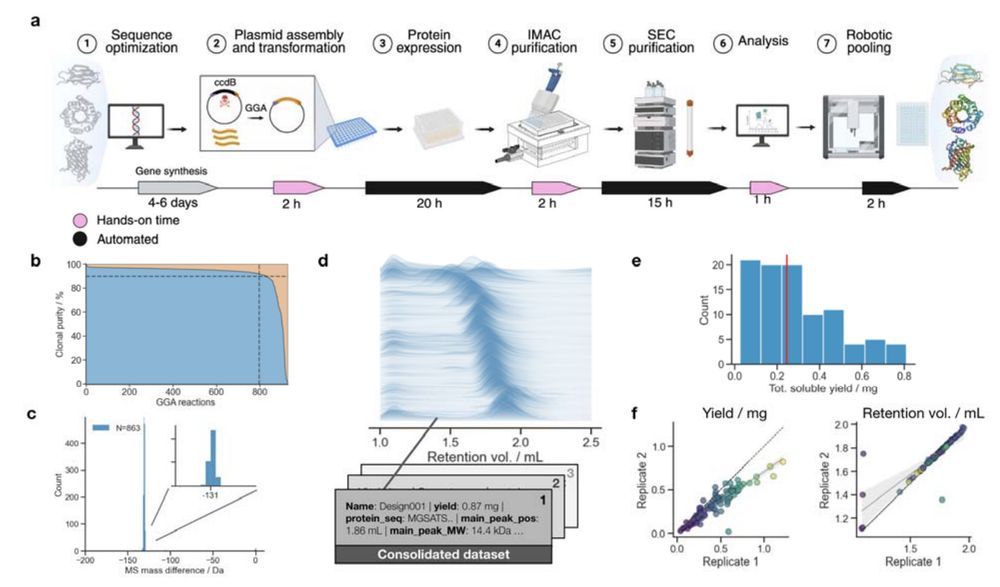
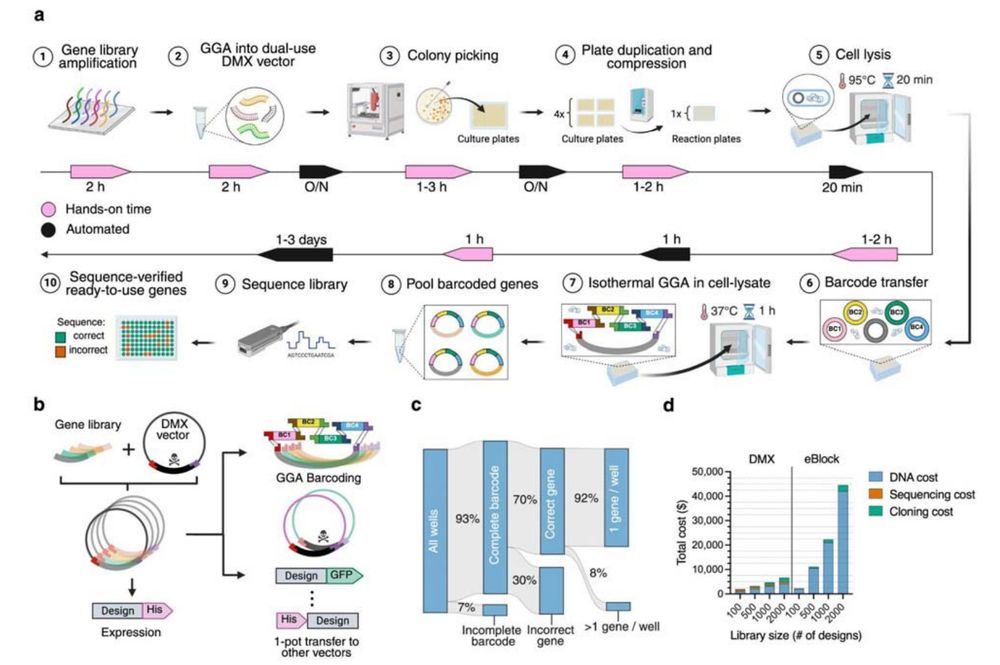
August 12, 2025 at 2:34 PM
The @uwproteindesign.bsky.social's experimental pipeline behind models like RFdiffusion and ProteinMPNN:
- A rapid, scalable, pipeline for producing and characterizing proteins
- A demultiplexing protocol for converting oligopools to clonal constructs
Jason Qian @lfmilles.bsky.social Basile Wicky
- A rapid, scalable, pipeline for producing and characterizing proteins
- A demultiplexing protocol for converting oligopools to clonal constructs
Jason Qian @lfmilles.bsky.social Basile Wicky
A benchmark dataset of 614 experimentally characterized de novo designed monomers from 11 different design studies shows that:
- deep learning structural metrics only weakly predict success
- The score distribution is different for different types of structures
@grocklin.bsky.social
- deep learning structural metrics only weakly predict success
- The score distribution is different for different types of structures
@grocklin.bsky.social
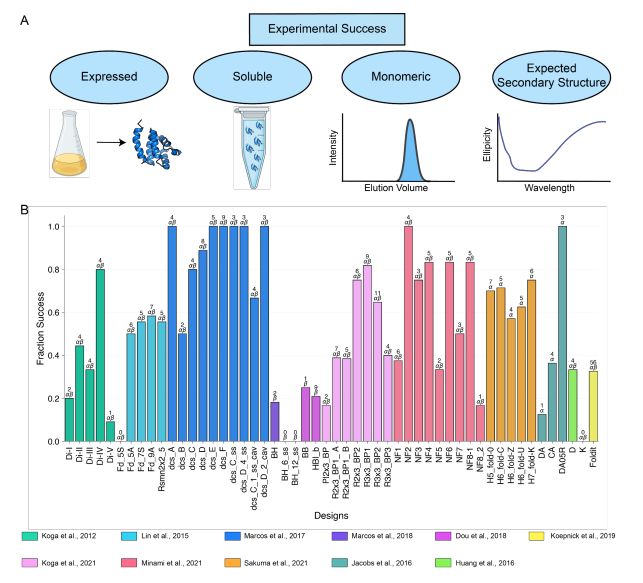



August 8, 2025 at 8:10 PM
A benchmark dataset of 614 experimentally characterized de novo designed monomers from 11 different design studies shows that:
- deep learning structural metrics only weakly predict success
- The score distribution is different for different types of structures
@grocklin.bsky.social
- deep learning structural metrics only weakly predict success
- The score distribution is different for different types of structures
@grocklin.bsky.social
MSA Pairformer efficiently extracts structure, protein-protein interactions, and mutation effects from MSAs by decomposing the effects of phylogeny and structural contacts.
@yoakiyama.bsky.social Zhidian Zhang @milot.bsky.social @martinsteinegger.bsky.social @sokrypton.org
@yoakiyama.bsky.social Zhidian Zhang @milot.bsky.social @martinsteinegger.bsky.social @sokrypton.org



August 5, 2025 at 9:17 PM
MSA Pairformer efficiently extracts structure, protein-protein interactions, and mutation effects from MSAs by decomposing the effects of phylogeny and structural contacts.
@yoakiyama.bsky.social Zhidian Zhang @milot.bsky.social @martinsteinegger.bsky.social @sokrypton.org
@yoakiyama.bsky.social Zhidian Zhang @milot.bsky.social @martinsteinegger.bsky.social @sokrypton.org
I just want to be a good area chair
August 4, 2025 at 6:43 PM
I just want to be a good area chair
- Model bacterial genomes as sequences of proteins
- predict protein-protein interactions, operon structure, and protein function
- infer phenotypic traits
- design synthetic genomes with desired properties
@macwiatrak.bsky.social @mariabrbic.bsky.social
@andresfloto.bsky.social
- predict protein-protein interactions, operon structure, and protein function
- infer phenotypic traits
- design synthetic genomes with desired properties
@macwiatrak.bsky.social @mariabrbic.bsky.social
@andresfloto.bsky.social
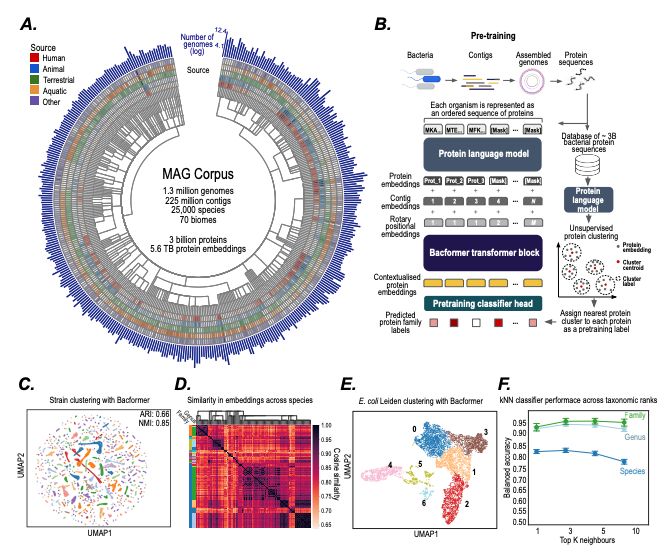
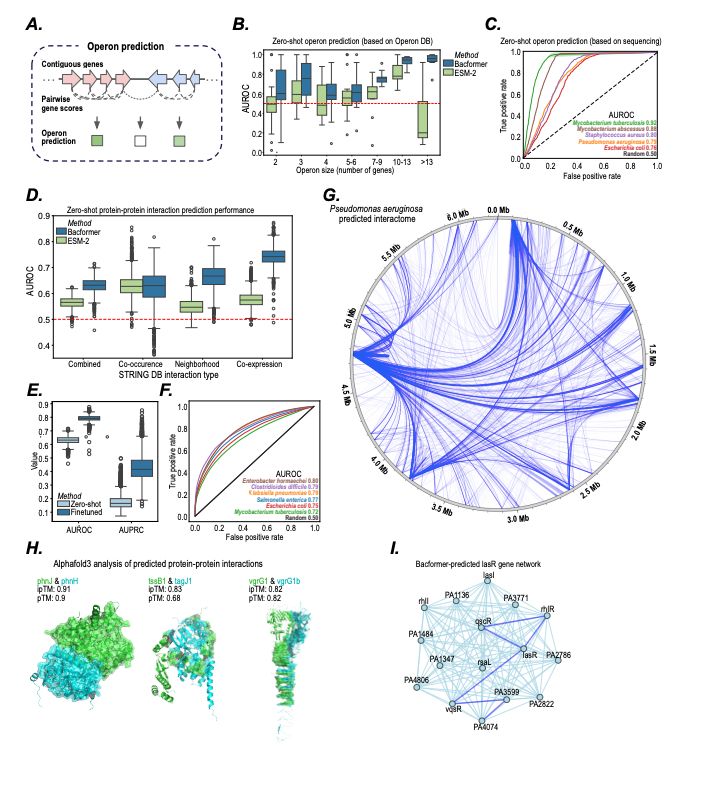
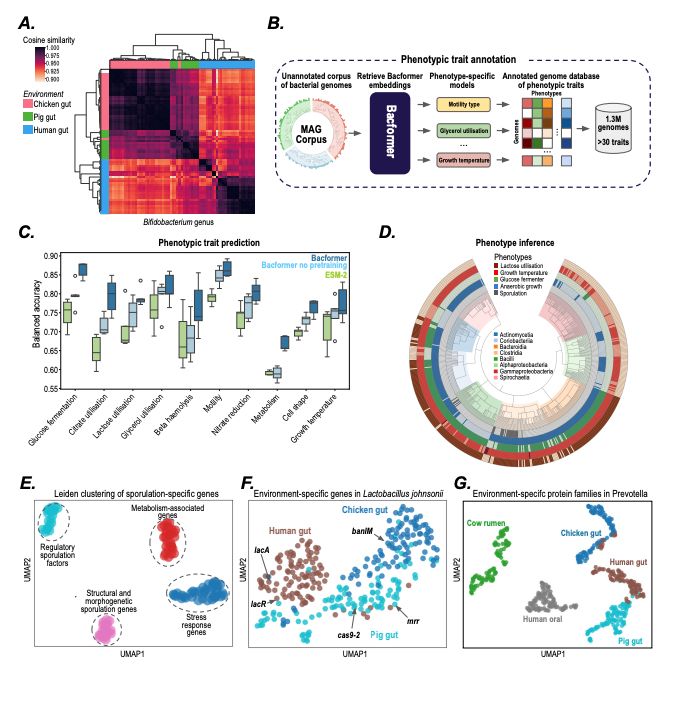
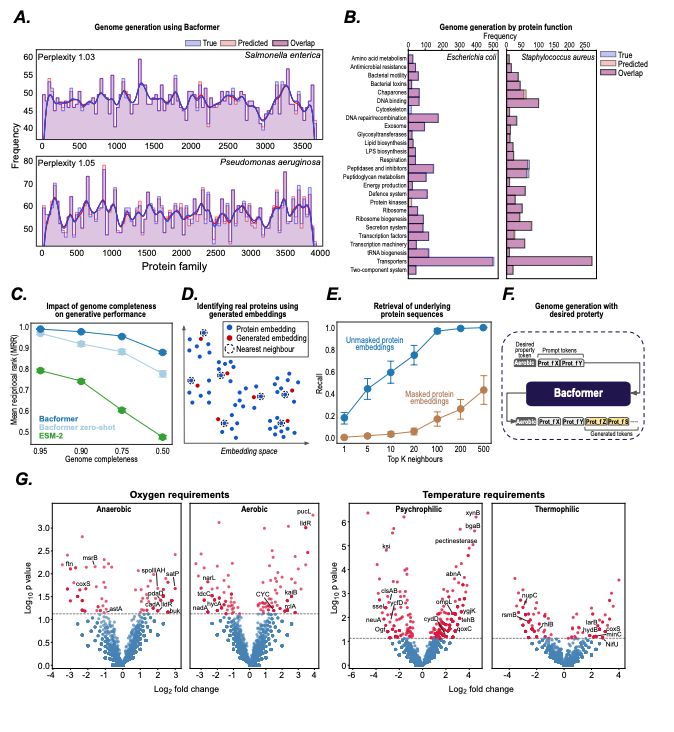
July 31, 2025 at 8:25 PM
- Model bacterial genomes as sequences of proteins
- predict protein-protein interactions, operon structure, and protein function
- infer phenotypic traits
- design synthetic genomes with desired properties
@macwiatrak.bsky.social @mariabrbic.bsky.social
@andresfloto.bsky.social
- predict protein-protein interactions, operon structure, and protein function
- infer phenotypic traits
- design synthetic genomes with desired properties
@macwiatrak.bsky.social @mariabrbic.bsky.social
@andresfloto.bsky.social
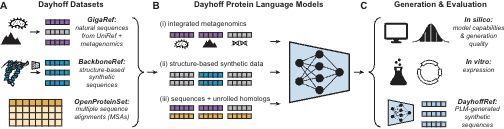
Learning on GigaRef yielded a small increase in the fraction of expressed proteins. Increasing model and dataset scale further improved the expression rate. Augmenting training with structure-based synthetic data from BackboneRef produced the highest expression success rate.

July 25, 2025 at 10:05 PM
Learning on GigaRef yielded a small increase in the fraction of expressed proteins. Increasing model and dataset scale further improved the expression rate. Augmenting training with structure-based synthetic data from BackboneRef produced the highest expression success rate.
How do dataset choice and model scale affect the quality of proteins generated by the Dayhoff model? In the first study of its kind, we generated sequences from different Dayhoff models and tested them head-to-head in the lab, measuring whether expressed in E. coli.
July 25, 2025 at 10:05 PM
How do dataset choice and model scale affect the quality of proteins generated by the Dayhoff model? In the first study of its kind, we generated sequences from different Dayhoff models and tested them head-to-head in the lab, measuring whether expressed in E. coli.
The Dayhoff Atlas dramatically expands the scale and diversity of publicly available protein data by providing the largest open dataset of natural proteins to date, GigaRef, and a first-in-class, large-scale dataset of synthetic proteins, BackboneRef.


July 25, 2025 at 10:05 PM
The Dayhoff Atlas dramatically expands the scale and diversity of publicly available protein data by providing the largest open dataset of natural proteins to date, GigaRef, and a first-in-class, large-scale dataset of synthetic proteins, BackboneRef.
In 1965, Margaret Dayhoff published the Atlas of Protein Sequence and Structure, which collated the 65 proteins whose amino acid sequences were then known.
Inspired by that Atlas, today we are releasing the Dayhoff Atlas of protein sequence data and protein language models.
Inspired by that Atlas, today we are releasing the Dayhoff Atlas of protein sequence data and protein language models.
July 25, 2025 at 10:05 PM
In 1965, Margaret Dayhoff published the Atlas of Protein Sequence and Structure, which collated the 65 proteins whose amino acid sequences were then known.
Inspired by that Atlas, today we are releasing the Dayhoff Atlas of protein sequence data and protein language models.
Inspired by that Atlas, today we are releasing the Dayhoff Atlas of protein sequence data and protein language models.
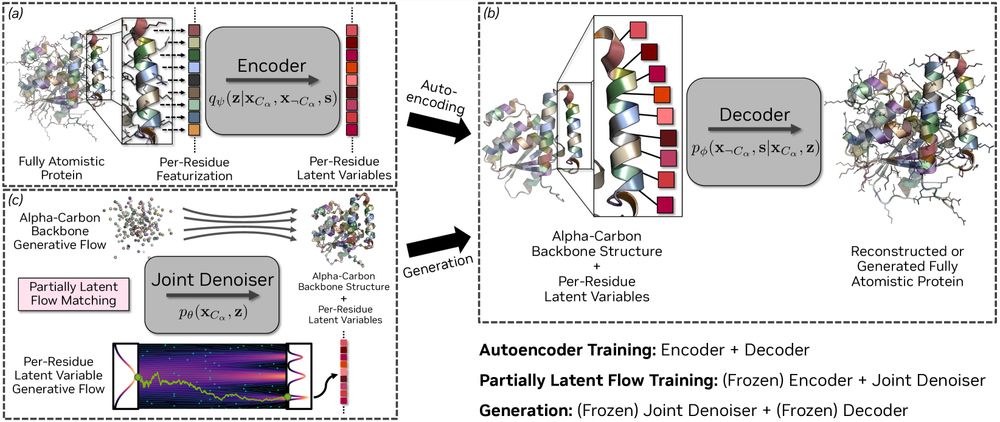
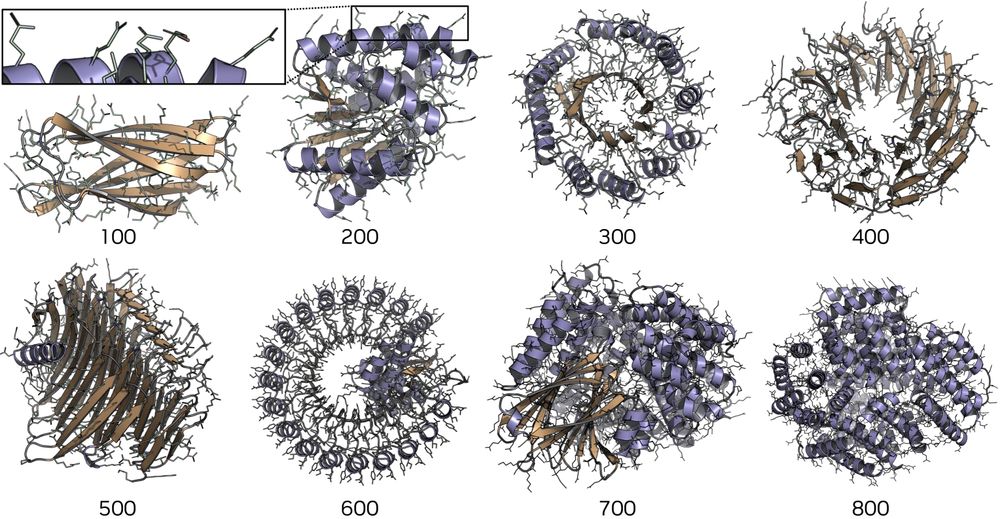
Partially-latent flow matching enables sequence-structure codesign of large proteins and functional motif scaffolding.
@kdidi.bsky.social @machine.learning.bio @karstenkreis.bsky.social @arashv.bsky.social
arxiv.org/html/2507.09...
@kdidi.bsky.social @machine.learning.bio @karstenkreis.bsky.social @arashv.bsky.social
arxiv.org/html/2507.09...

July 16, 2025 at 7:05 PM
Partially-latent flow matching enables sequence-structure codesign of large proteins and functional motif scaffolding.
@kdidi.bsky.social @machine.learning.bio @karstenkreis.bsky.social @arashv.bsky.social
arxiv.org/html/2507.09...
@kdidi.bsky.social @machine.learning.bio @karstenkreis.bsky.social @arashv.bsky.social
arxiv.org/html/2507.09...
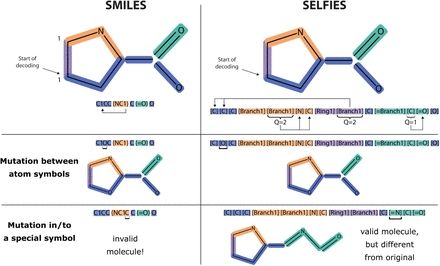
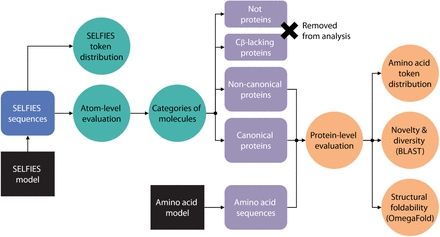

June 25, 2025 at 2:19 PM
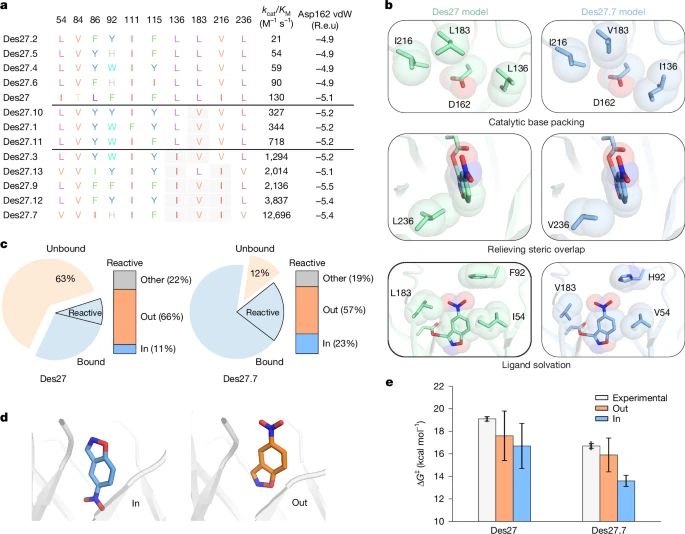
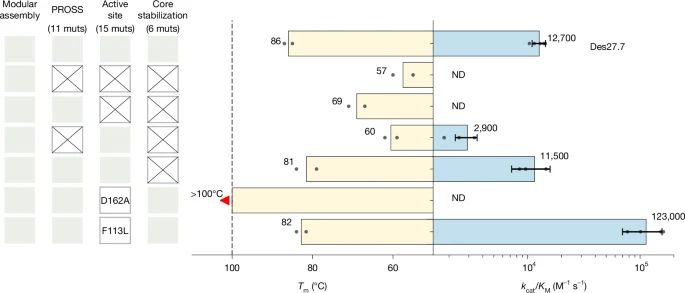
Physics-based design of efficient Kemp eliminases
@lynnkamerlin.bsky.social
www.nature.com/articles/s41...
@lynnkamerlin.bsky.social
www.nature.com/articles/s41...
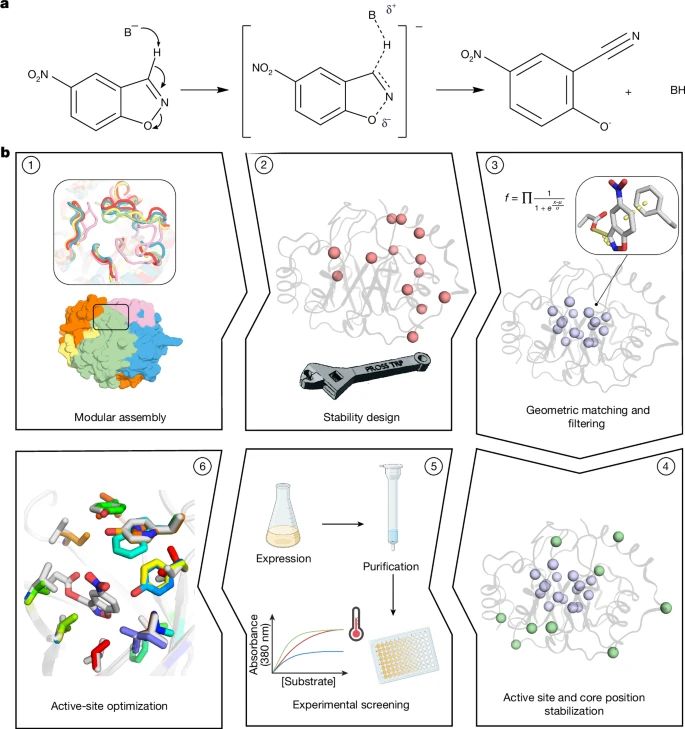
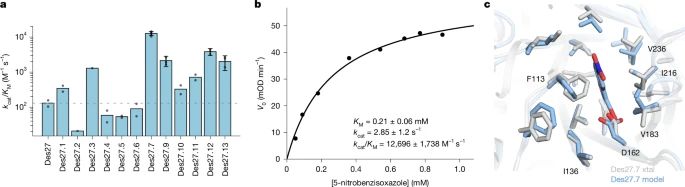
June 19, 2025 at 9:18 PM
Physics-based design of efficient Kemp eliminases
@lynnkamerlin.bsky.social
www.nature.com/articles/s41...
@lynnkamerlin.bsky.social
www.nature.com/articles/s41...
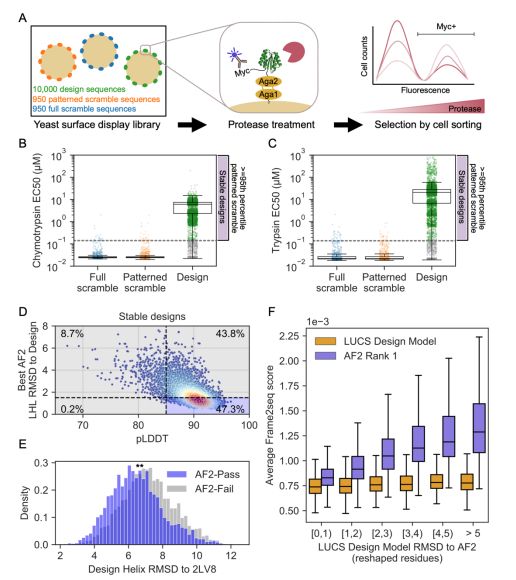
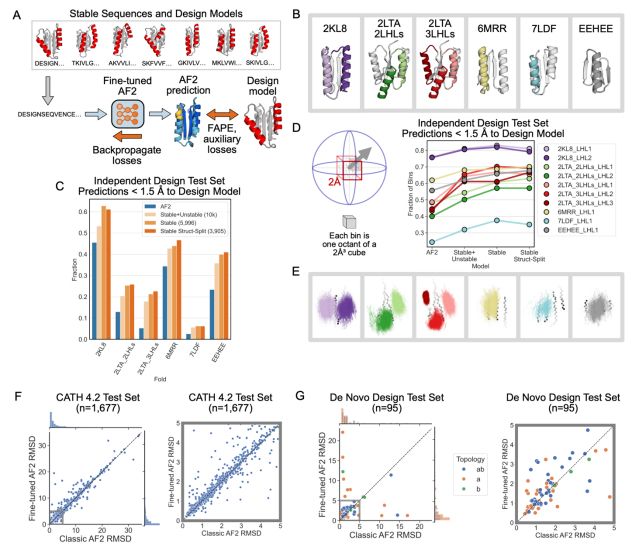
Deep learning methods for protein structure prediction and design produce idealized structures. Finetuning on a set of physics-based de novo proteins improves their geometric diversity and generalization capabilities.
@benorr.bsky.social @kortemmelab.bsky.social
www.biorxiv.org/content/10.1...
@benorr.bsky.social @kortemmelab.bsky.social
www.biorxiv.org/content/10.1...
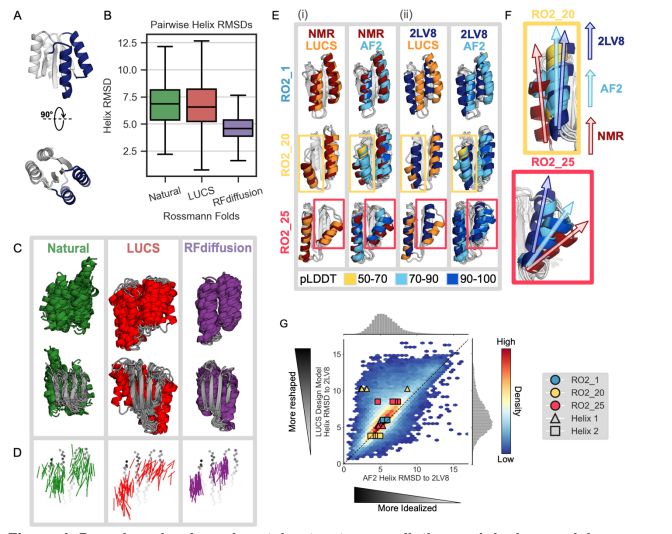
June 16, 2025 at 6:39 PM
Deep learning methods for protein structure prediction and design produce idealized structures. Finetuning on a set of physics-based de novo proteins improves their geometric diversity and generalization capabilities.
@benorr.bsky.social @kortemmelab.bsky.social
www.biorxiv.org/content/10.1...
@benorr.bsky.social @kortemmelab.bsky.social
www.biorxiv.org/content/10.1...