



Jose Rebolledo-Oyarce
@jrebolledoo.bsky.social
PhD Student at University of Notre Dame | Computational Chemistry | Heterogeneous Catalysis
Reposted by Jose Rebolledo-Oyarce

Call for Applications 2025: Lise Meitner #research Groups
https://www.mpg.de/lise-meitner-excellence-program
Programme by the @maxplanckgesellschaft designed to attract and specifically promote exceptionally qualified female #scientists
deadline April 08th, 2025.
note that you should contact […]
https://www.mpg.de/lise-meitner-excellence-program
Programme by the @maxplanckgesellschaft designed to attract and specifically promote exceptionally qualified female #scientists
deadline April 08th, 2025.
note that you should contact […]
Original post on fediscience.org
fediscience.org
February 12, 2025 at 6:50 PM
Call for Applications 2025: Lise Meitner #research Groups
https://www.mpg.de/lise-meitner-excellence-program
Programme by the @maxplanckgesellschaft designed to attract and specifically promote exceptionally qualified female #scientists
deadline April 08th, 2025.
note that you should contact […]
https://www.mpg.de/lise-meitner-excellence-program
Programme by the @maxplanckgesellschaft designed to attract and specifically promote exceptionally qualified female #scientists
deadline April 08th, 2025.
note that you should contact […]
Reposted by Jose Rebolledo-Oyarce

Out now in Nature Human Behaviour: Our 68-country #survey on public attitudes to #science 📣
It shows: People still #trust scientists and support an active role of scientists in society and policy-making. #OpenAccess available here: www.nature.com/articles/s41... @natureportfolio.bsky.social
(1/13)
It shows: People still #trust scientists and support an active role of scientists in society and policy-making. #OpenAccess available here: www.nature.com/articles/s41... @natureportfolio.bsky.social
(1/13)

January 20, 2025 at 10:27 AM
Out now in Nature Human Behaviour: Our 68-country #survey on public attitudes to #science 📣
It shows: People still #trust scientists and support an active role of scientists in society and policy-making. #OpenAccess available here: www.nature.com/articles/s41... @natureportfolio.bsky.social
(1/13)
It shows: People still #trust scientists and support an active role of scientists in society and policy-making. #OpenAccess available here: www.nature.com/articles/s41... @natureportfolio.bsky.social
(1/13)
Reposted by Jose Rebolledo-Oyarce

Lots of new folks here on BlueSky, so perhaps time for a (re)introduction! I'm Evan (they/them), and I'm a chemical researcher. Currently a postdoc at CMU, soon to be an assistant prof. in chemical engineering. #ChemSky #CompChem #AcademicSky #LGBTQSTEM #DisabledInSTEM 🧪 1/n
October 21, 2024 at 5:16 PM
Lots of new folks here on BlueSky, so perhaps time for a (re)introduction! I'm Evan (they/them), and I'm a chemical researcher. Currently a postdoc at CMU, soon to be an assistant prof. in chemical engineering. #ChemSky #CompChem #AcademicSky #LGBTQSTEM #DisabledInSTEM 🧪 1/n
Reposted by Jose Rebolledo-Oyarce

Active-learning ML approach for catalytic free-energy calculations by Liu et al.: An MLP “committee” and constrained MD refine data, capturing reaction pathways 10^6× faster than ab initio. Their scheme ensures robust coverage of transitions and near-DFT precision. pubs.acs.org/doi/10.1021/...
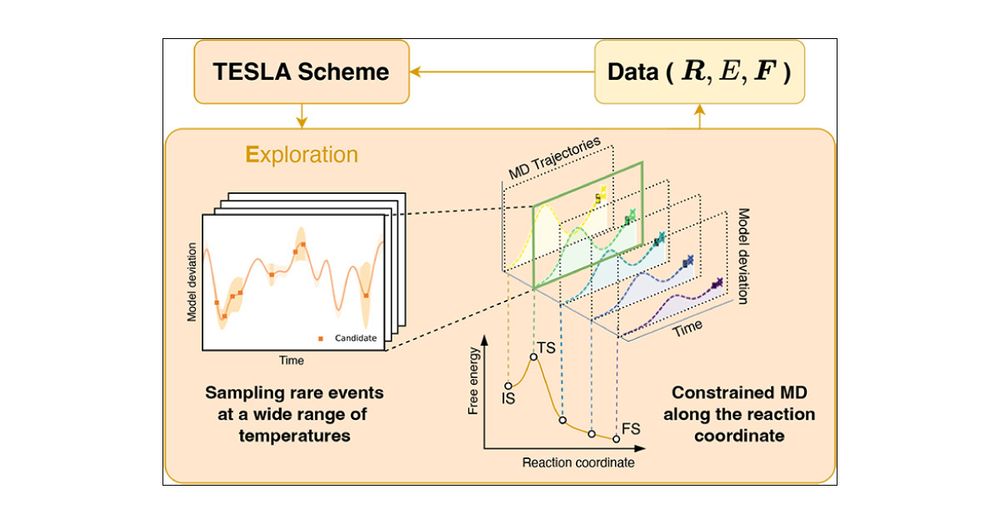
CatFlow: An Automated Workflow for Training Machine Learning Potentials to Compute Free Energies in Dynamic Catalysis
Dynamic effects of catalysts play a crucial role in catalytic reactions, necessitating the incorporation of statistical sampling and understanding of the impact of dynamic structures in free energy calculations. However, the complexity of catalytic systems poses challenges in effectively exploring the vast configurational space effectively. In this work, we propose CatFlow, an automated workflow for training machine learning potentials (MLPs) to compute free energies of catalytic reactions. CatFlow combines constrained molecular dynamics (MD) simulation with concurrent training of MLPs and sequential calculation of free energies with well trained MLPs. By rapidly generating reliable MLPs, CatFlow facilitates rigorous free energy calculations, enabling the determination of the reaction profiles in an end-to-end manner. We showcased the capabilities of CatFlow by investigating the activation of O2 catalyzed by Pt clusters and demonstrated the effects of phase transition on the activities of the catalytic reaction. CatFlow offers an efficient and automated solution for studying the catalytic elementary reaction processes. It reduces the need for human intervention and provides researchers with a powerful tool to investigate free energies of dynamic catalysis.
pubs.acs.org
January 1, 2025 at 5:08 PM
Active-learning ML approach for catalytic free-energy calculations by Liu et al.: An MLP “committee” and constrained MD refine data, capturing reaction pathways 10^6× faster than ab initio. Their scheme ensures robust coverage of transitions and near-DFT precision. pubs.acs.org/doi/10.1021/...
Reposted by Jose Rebolledo-Oyarce

Online Symposium on Molecular Machine Learning on January 16th, organized by the Glorius Group at @unimuenster.bsky.social - with @fjduarte.bsky.social, @kulikgroup.bsky.social, Bingqing Cheng, Matthew Sigman: www.uni-muenster.de/Chemie.oc/gl.... Registration is free and highly recommended!

December 17, 2024 at 3:48 PM
Online Symposium on Molecular Machine Learning on January 16th, organized by the Glorius Group at @unimuenster.bsky.social - with @fjduarte.bsky.social, @kulikgroup.bsky.social, Bingqing Cheng, Matthew Sigman: www.uni-muenster.de/Chemie.oc/gl.... Registration is free and highly recommended!
Reposted by Jose Rebolledo-Oyarce

@deepmind.google.web.brid.gy published a really cool method for predicting crystals that can be synthesised from amorphous precursors. They construct candidate cells by sampling geometries from a modeled amorphous cells and then optimising those cells.
#compchem
www.nature.com/articles/s43...
#compchem
www.nature.com/articles/s43...

Predicting emergence of crystals from amorphous precursors with deep learning potentials - Nature Computational Science
This study introduces a2c, a computational method that leverages machine learning and atomistic simulations to predict the most likely crystallization products upon annealing of amorphous precursors. ...
www.nature.com
December 19, 2024 at 10:54 AM
@deepmind.google.web.brid.gy published a really cool method for predicting crystals that can be synthesised from amorphous precursors. They construct candidate cells by sampling geometries from a modeled amorphous cells and then optimising those cells.
#compchem
www.nature.com/articles/s43...
#compchem
www.nature.com/articles/s43...
Reposted by Jose Rebolledo-Oyarce

📢 It's our great pleasure to invite you all to our upcoming #Workshop on "Computer Simulation and #Theory of Macromolecules" in #Hünfeld on March 7-8, 2025 in #hybrid format! See you online or on-site! More details are provided here: www.mpinat.mpg.de/workshop/hue... #TheoryHünfeld2025 #biophysics

December 4, 2024 at 1:43 PM
📢 It's our great pleasure to invite you all to our upcoming #Workshop on "Computer Simulation and #Theory of Macromolecules" in #Hünfeld on March 7-8, 2025 in #hybrid format! See you online or on-site! More details are provided here: www.mpinat.mpg.de/workshop/hue... #TheoryHünfeld2025 #biophysics
Reposted by Jose Rebolledo-Oyarce

Great chance to work with my friend Esteban Vöhringer-Martinez in Chile 🇨🇱 qcmm.udec.cl/people/prof-...

December 4, 2024 at 3:36 PM
Great chance to work with my friend Esteban Vöhringer-Martinez in Chile 🇨🇱 qcmm.udec.cl/people/prof-...
Reposted by Jose Rebolledo-Oyarce

Multiple PhD positions available
in our group in the field of computational and theoretical chemistry
Feel free to forward the info and please apply via
phdheidelberg@gmail.com
in our group in the field of computational and theoretical chemistry
Feel free to forward the info and please apply via
phdheidelberg@gmail.com
December 4, 2024 at 4:54 PM
Multiple PhD positions available
in our group in the field of computational and theoretical chemistry
Feel free to forward the info and please apply via
phdheidelberg@gmail.com
in our group in the field of computational and theoretical chemistry
Feel free to forward the info and please apply via
phdheidelberg@gmail.com
Reposted by Jose Rebolledo-Oyarce

I created a starter pack for chemical and biomolecular engineers! Let me know if you want to be added to the list #chemE go.bsky.app/DLGAANj
November 22, 2024 at 4:41 PM
I created a starter pack for chemical and biomolecular engineers! Let me know if you want to be added to the list #chemE go.bsky.app/DLGAANj
Reposted by Jose Rebolledo-Oyarce

If you want to help your students move from just writing code and scripts to actually developing scientific software that is useable and maintainable in the long-term, check out our latest paper in JCE. pubs.acs.org/doi/10.1021/...

Cookiecutter for Computational Molecular Sciences: A Best Practices Ready Python Project Generator
Scientific software development takes far more than good programming abilities and scientific reasoning. Concepts such as version control, continuous integration, packaging, deployment, automatic documentation compiling, licensing, and even file structure are not traditionally taught to scientific programmers. The skill gap leads to inconsistent code quality and difficulty deploying products to the broader audience. Most of the implementation of these skills however can be constructed at project inception. The Cookiecutter for Computational Molecular Sciences generates ready-to-go Python projects that incorporate all of the concepts above from a single command. The final product is then a software project which lets developers focus on the science and minimizes worries about nonscientific and nonprogramming concepts because the best practices, as established by the Molecular Sciences Software Institute, have already been incorporated for them. This is a community driven project with widespread adoption across the computational molecular sciences. The Molecular Sciences Software Institute and Computational Molecular Sciences community also continually contribute and update the Cookiecutter for Computational Molecular Science, ensuring that the project is responsive to community needs and tool updates. All are welcome to suggest changes and contribute to making this the best starting point for Python-based scientific code.
pubs.acs.org
November 16, 2024 at 10:08 PM
If you want to help your students move from just writing code and scripts to actually developing scientific software that is useable and maintainable in the long-term, check out our latest paper in JCE. pubs.acs.org/doi/10.1021/...
Reposted by Jose Rebolledo-Oyarce

Hi #ChemSky - recent Starter Pack for our #GreenChemistry and #SustainableChemistry community out there! Please give it a follow and let me know if you'd like to be included! 🌱🌎
go.bsky.app/Bmp4DHn
go.bsky.app/Bmp4DHn
November 13, 2024 at 4:59 PM
Hi #ChemSky - recent Starter Pack for our #GreenChemistry and #SustainableChemistry community out there! Please give it a follow and let me know if you'd like to be included! 🌱🌎
go.bsky.app/Bmp4DHn
go.bsky.app/Bmp4DHn
Reposted by Jose Rebolledo-Oyarce

go.bsky.app/Qc4frbt
Made a starter pack for computational chemists, let me know who I missed please
@jchodera.bsky.social
@olexandr.bsky.social
@jelfschem.bsky.social
Made a starter pack for computational chemists, let me know who I missed please
@jchodera.bsky.social
@olexandr.bsky.social
@jelfschem.bsky.social
November 12, 2024 at 2:17 PM
go.bsky.app/Qc4frbt
Made a starter pack for computational chemists, let me know who I missed please
@jchodera.bsky.social
@olexandr.bsky.social
@jelfschem.bsky.social
Made a starter pack for computational chemists, let me know who I missed please
@jchodera.bsky.social
@olexandr.bsky.social
@jelfschem.bsky.social