




Jeff Vierstra
@jeffvierstra.bsky.social
Senior Investigator @ Altius Institute for Biomedical Sciences. Research: High-resolution mapping of chromatin structure & function. Fun: Mountain shenanigans and skiing turns all year. Seattle, USA/Patagonia Chilena (🇺🇸🇨🇱). http://vierstra.org
Double full rainbow at the end of the world at Cape Horn, Chile 🇨🇱

November 12, 2025 at 3:11 PM
Double full rainbow at the end of the world at Cape Horn, Chile 🇨🇱
With all the wild stuff going on in the States (and the world) I am going to escape reality for a while on a sailing trip to the end of the world around Cape Horn and the Beagle Channel (named after the HMS Beagle of Charles Darwin and Robert Fitzroy fame). Thinking this might be type 2 fun...

November 8, 2025 at 12:53 AM
With all the wild stuff going on in the States (and the world) I am going to escape reality for a while on a sailing trip to the end of the world around Cape Horn and the Beagle Channel (named after the HMS Beagle of Charles Darwin and Robert Fitzroy fame). Thinking this might be type 2 fun...
Southern fjords of Chile on a boat.

October 29, 2025 at 11:17 AM
Southern fjords of Chile on a boat.
Does one sample (or even 10) suffice to define core cell type regulatory elements? NO! Because of both biological and technical variability you need to profile many (typically >15). The additional peaks are enriched for trait associated variants, so you miss a lot of possibly important signal.

August 29, 2025 at 8:32 PM
Does one sample (or even 10) suffice to define core cell type regulatory elements? NO! Because of both biological and technical variability you need to profile many (typically >15). The additional peaks are enriched for trait associated variants, so you miss a lot of possibly important signal.
Look at this and tell me I am wrong : DNaseI footprinting data is unparalleled in genomics. ~700 high quality datasets for an upcoming ENCODE data drop.

August 20, 2025 at 11:22 PM
Look at this and tell me I am wrong : DNaseI footprinting data is unparalleled in genomics. ~700 high quality datasets for an upcoming ENCODE data drop.
Activity determining nucleotides on the BCL11A +58 enhancer according to a ML model built purely on DNase I data from thousands of cell types (this is just prediction for erythroid cells). Not bad w.r.t. functional data. The GATA1 site is the therapeutic target of Casgevy for SCD and B-thal.

August 13, 2025 at 9:36 PM
Activity determining nucleotides on the BCL11A +58 enhancer according to a ML model built purely on DNase I data from thousands of cell types (this is just prediction for erythroid cells). Not bad w.r.t. functional data. The GATA1 site is the therapeutic target of Casgevy for SCD and B-thal.
You might know that my life mostly revolves around skiing. I am organizing a 25 day sail & ski trip to Antarctica in Dec. 2025 and have space for 1-2 more people. We leave from Ushuaia, AR on the Tierra del Fuego (early Dec.) DM me for details and pass this around if you know anyone interested!


July 12, 2025 at 3:09 AM
You might know that my life mostly revolves around skiing. I am organizing a 25 day sail & ski trip to Antarctica in Dec. 2025 and have space for 1-2 more people. We leave from Ushuaia, AR on the Tierra del Fuego (early Dec.) DM me for details and pass this around if you know anyone interested!
Here is slide demonstrating how it works w.r.t. to the highly aneuploid, yet very commonly used cell-line K562.

July 8, 2025 at 6:06 PM
Here is slide demonstrating how it works w.r.t. to the highly aneuploid, yet very commonly used cell-line K562.
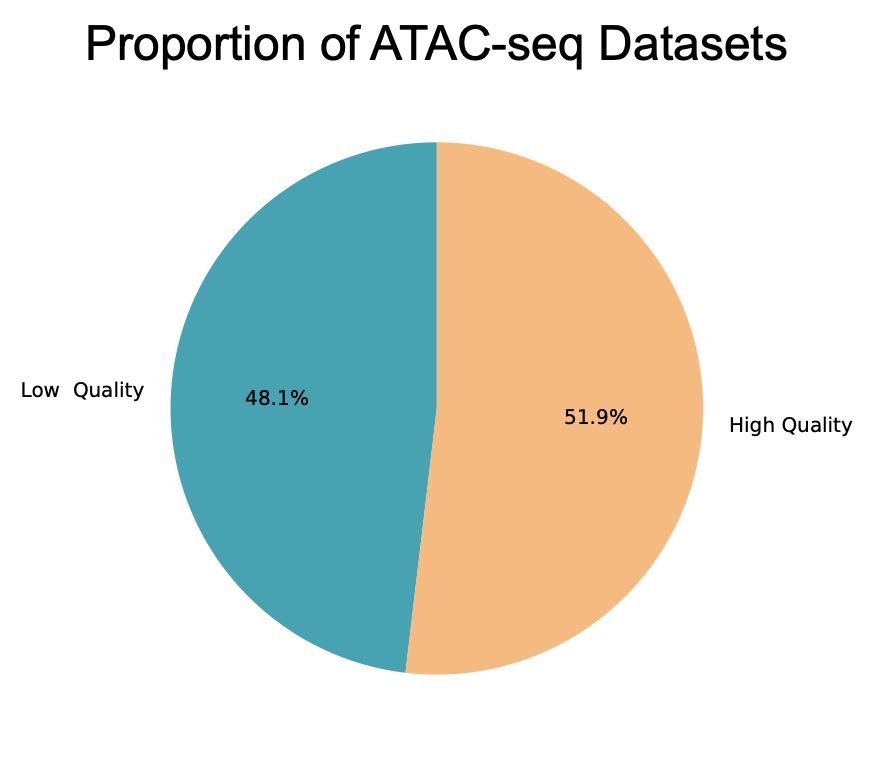
PSA: We just finished processing nearly all public ATAC-seq datasets from SRA (about 22,000 datasets). (Not?) Surprisingly, we had to throw-out nearly ~50% because they were low-quality (low signal-to-noise, duplicate rate, etc.). Check quality before analysis (TSS-enrichment is not sufficient!).
May 13, 2025 at 7:42 PM
PSA: We just finished processing nearly all public ATAC-seq datasets from SRA (about 22,000 datasets). (Not?) Surprisingly, we had to throw-out nearly ~50% because they were low-quality (low signal-to-noise, duplicate rate, etc.). Check quality before analysis (TSS-enrichment is not sufficient!).
Two genome scientists at the end of the world!

May 12, 2025 at 4:42 PM
Two genome scientists at the end of the world!
Screening is hard in primary cells given the considerable positional effects when using lentivirus. We estimate that you need roughly 800 integrations per sequence accurately estimate effect size. This hard to do in prim. HSPCs in which 5M is a lot cells, and really limits you input library size.

May 9, 2025 at 3:32 PM
Screening is hard in primary cells given the considerable positional effects when using lentivirus. We estimate that you need roughly 800 integrations per sequence accurately estimate effect size. This hard to do in prim. HSPCs in which 5M is a lot cells, and really limits you input library size.
I think this study exposes a well-known yet often ignored problem -- cell-context matters. Screening erythroid candidate sequences in a cancer line (K562) gives completely different results than primary cells. In fact we would have discarded these sequences if only measured in K562.

May 9, 2025 at 3:24 PM
I think this study exposes a well-known yet often ignored problem -- cell-context matters. Screening erythroid candidate sequences in a cancer line (K562) gives completely different results than primary cells. In fact we would have discarded these sequences if only measured in K562.


Hmm. I wonder if they will change what constitutes allowable indirect and direct expeditures. Will individual labs be able to negotiate lab space (i.e., rent)?

February 20, 2025 at 3:07 AM
Hmm. I wonder if they will change what constitutes allowable indirect and direct expeditures. Will individual labs be able to negotiate lab space (i.e., rent)?
In this case it's simple: it's predicting the creation of a weak GATA1::TAL1 motif. If the model came from K562 it's not surprising given the prevalence of this motif in that cell type.

February 13, 2025 at 3:20 AM
In this case it's simple: it's predicting the creation of a weak GATA1::TAL1 motif. If the model came from K562 it's not surprising given the prevalence of this motif in that cell type.
I find the current administration abhorrent, and this will be painful, and it's not how I would do it. We must recognize that we have been living unsustainably years. Here is an interesting document from the center-left Progressive Policy Institute (www.progressivepolicy.org/wp-content/u...).

February 8, 2025 at 2:34 AM
I find the current administration abhorrent, and this will be painful, and it's not how I would do it. We must recognize that we have been living unsustainably years. Here is an interesting document from the center-left Progressive Policy Institute (www.progressivepolicy.org/wp-content/u...).
We have a new peak caller that simultaneously calls large CNVs from DNaseI/ATC data. No cell (not even primary) are completely normal, but one can quantify/estimate how many non-diploid blocks a genome has and then sort datasets by this. In the plot below, 1 is normal, 2 is on average triploid.

January 29, 2025 at 5:29 AM
We have a new peak caller that simultaneously calls large CNVs from DNaseI/ATC data. No cell (not even primary) are completely normal, but one can quantify/estimate how many non-diploid blocks a genome has and then sort datasets by this. In the plot below, 1 is normal, 2 is on average triploid.
>90% of ENCODE chromatin accessibility data is from normal tissue. Like 3000 datasets covering >350 cell types/states/etc.

January 29, 2025 at 4:25 AM
>90% of ENCODE chromatin accessibility data is from normal tissue. Like 3000 datasets covering >350 cell types/states/etc.
Cancer cells are actually kind of interesting in their own right because they sort of reactivate chromatin used in primitive or other lineages. Here is a little teaser image from a paper we are putting together.

January 28, 2025 at 1:38 AM
Cancer cells are actually kind of interesting in their own right because they sort of reactivate chromatin used in primitive or other lineages. Here is a little teaser image from a paper we are putting together.
I just read the recent "Multiscale footprints reveal..." paper. (www.nature.com/articles/s41...). The claim by authors (below) is based on ONLY 2 regions (amongst 100k) in 5 cell types. They don't provide genome-wide footprint calls, so how can they make these claims?! i.e., where's the evidence?

January 25, 2025 at 11:39 PM
I just read the recent "Multiscale footprints reveal..." paper. (www.nature.com/articles/s41...). The claim by authors (below) is based on ONLY 2 regions (amongst 100k) in 5 cell types. They don't provide genome-wide footprint calls, so how can they make these claims?! i.e., where's the evidence?
2. We (and others) have shown that you can correct for this with principled statistics. Our recent (4yrs ago) footprinting paper described this, but we didn't give it our method a fancy name, so maybe people didn't realize/appreciate the technical advances made. www.nature.com/articles/s41... 3/n

January 24, 2025 at 10:54 PM
2. We (and others) have shown that you can correct for this with principled statistics. Our recent (4yrs ago) footprinting paper described this, but we didn't give it our method a fancy name, so maybe people didn't realize/appreciate the technical advances made. www.nature.com/articles/s41... 3/n
Also missing some references -- looking at data at different scales is not new -- we published a similar idea ten years ago! Simply binning fragments by size gets you 95% there.


January 23, 2025 at 7:33 PM
Also missing some references -- looking at data at different scales is not new -- we published a similar idea ten years ago! Simply binning fragments by size gets you 95% there.
Going back to my hometown (Madison, WI) in January reminds me of why I live in the PNW now. Good thing I still own fleece lined pants!

January 17, 2025 at 5:08 PM
Going back to my hometown (Madison, WI) in January reminds me of why I live in the PNW now. Good thing I still own fleece lined pants!