



Ezequiel Galpern
@eag91.bsky.social
MSCA fellow at @crg.eu w/M. Dias and @jonnyfrazer.bsky.social
> Biological Physics | Proteins | Comp Bio | ML
https://scholar.google.com/citations?user=n55NtEsAAAAJ&hl=en
> Biological Physics | Proteins | Comp Bio | ML
https://scholar.google.com/citations?user=n55NtEsAAAAJ&hl=en
Dark Energy is not a relative score, but provides a common scale and has physical energy units. And it can be also computed even if ΔΔG is not available, using AWSEM force-field. You can compute it for your favorite PDB colab.research.google.com/github/eagalpern/colabs/blob/main/DarkEnergy.ipynb

Google Colab
colab.research.google.com
August 12, 2025 at 2:49 PM
Dark Energy is not a relative score, but provides a common scale and has physical energy units. And it can be also computed even if ΔΔG is not available, using AWSEM force-field. You can compute it for your favorite PDB colab.research.google.com/github/eagalpern/colabs/blob/main/DarkEnergy.ipynb
Some sites have, on average over all the possible variants, high Dark Energy. In those cases, there is a function beyond protein folding affecting natural selection. This is clearly the case of Enzyme catalytic sites

August 12, 2025 at 2:49 PM
Some sites have, on average over all the possible variants, high Dark Energy. In those cases, there is a function beyond protein folding affecting natural selection. This is clearly the case of Enzyme catalytic sites
The idea is simple. For a deleterious mutation, we expect the protein to get destabilized. But how much? Correlations between a ΔΔG DMS and the corresponding ESM-2 scores are not perfect. We define the difference between these two free energies as a ‘Dark Energy’.

August 12, 2025 at 2:49 PM
The idea is simple. For a deleterious mutation, we expect the protein to get destabilized. But how much? Correlations between a ΔΔG DMS and the corresponding ESM-2 scores are not perfect. We define the difference between these two free energies as a ‘Dark Energy’.
And you can make folding mechanism predictions for your favorite protein!
colab.research.google.com/github/eagal...
colab.research.google.com/github/eagal...
Google Colab
colab.research.google.com
July 15, 2025 at 8:57 PM
And you can make folding mechanism predictions for your favorite protein!
colab.research.google.com/github/eagal...
colab.research.google.com/github/eagal...
Also, we show that both the stability and cooperativity changes induced by mutations can be computed directly using sequence-based evolutionary models.
6/n
6/n

July 15, 2025 at 8:57 PM
Also, we show that both the stability and cooperativity changes induced by mutations can be computed directly using sequence-based evolutionary models.
6/n
6/n
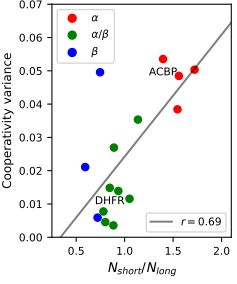
Protein topology imposes limits on the variability of folding cooperativity within a family. While most beta and alpha/beta structures exhibit only a few possible mechanisms despite high sequence diversity, alpha topologies allow for diverse folding scenarios.
5/n
5/n
July 15, 2025 at 8:57 PM
Protein topology imposes limits on the variability of folding cooperativity within a family. While most beta and alpha/beta structures exhibit only a few possible mechanisms despite high sequence diversity, alpha topologies allow for diverse folding scenarios.
5/n
5/n
For 15 diverse protein families, we computed the folding mechanisms of hundreds of proteins by simulating an Ising chain of folding elements, or foldons. The energetics is determined by each amino acid sequence.
4/n
4/n

July 15, 2025 at 8:57 PM
For 15 diverse protein families, we computed the folding mechanisms of hundreds of proteins by simulating an Ising chain of folding elements, or foldons. The energetics is determined by each amino acid sequence.
4/n
4/n
We show that it is possible to use sequence info to go beyond predicting native structures and global stability to infer the folding mechanisms of globular proteins. We mapped a Potts evolutionary energy at the amino-acid level to a coarse-grained description of folding.
3/n
3/n

July 15, 2025 at 8:57 PM
We show that it is possible to use sequence info to go beyond predicting native structures and global stability to infer the folding mechanisms of globular proteins. We mapped a Potts evolutionary energy at the amino-acid level to a coarse-grained description of folding.
3/n
3/n
We know that closely related proteins usually share similar three-dimensional structures. But differences in their amino acid sequences can lead to distinct folding mechanisms, enabling the natural evolution of diverse biological functions.
2/n
2/n
July 15, 2025 at 8:57 PM
We know that closely related proteins usually share similar three-dimensional structures. But differences in their amino acid sequences can lead to distinct folding mechanisms, enabling the natural evolution of diverse biological functions.
2/n
2/n