



Kartik Chundru
@chundru.bsky.social
Postdoc at University of Exeter 🇮🇪🇮🇳🇬🇧 Statistical/Computational analyses using any NGS-based data
Formerly at Sanger institute working on recessive developmental disorders in DDD
Formerly at Sanger institute working on recessive developmental disorders in DDD
Ha! Thanks, I'll file right under "Even reviewer 3 was speechless"
November 10, 2025 at 11:57 AM
Ha! Thanks, I'll file right under "Even reviewer 3 was speechless"
Thank you to other co-authors @carolinefwright.bsky.social, @mnweedon.bsky.social, @timfrayling.bsky.social, and @drarwood.bsky.social, @nihrexeterbrc.bsky.social, biobanks @ukbiobank.bsky.social and All of Us, and all of the participants of the studies
November 8, 2025 at 9:31 AM
Thank you to other co-authors @carolinefwright.bsky.social, @mnweedon.bsky.social, @timfrayling.bsky.social, and @drarwood.bsky.social, @nihrexeterbrc.bsky.social, biobanks @ukbiobank.bsky.social and All of Us, and all of the participants of the studies
A massive, massive thank you to @hiwwright.bsky.social, @rnbeaumont.bsky.social, @drghawkes.bsky.social who all really drove this project to completion. Without them I would still be twiddling my thumbs shouting to the clouds about QC (I still will, but now you can read about it too!)
November 8, 2025 at 9:31 AM
A massive, massive thank you to @hiwwright.bsky.social, @rnbeaumont.bsky.social, @drghawkes.bsky.social who all really drove this project to completion. Without them I would still be twiddling my thumbs shouting to the clouds about QC (I still will, but now you can read about it too!)
Using our DNANexus applet it is fast and not too expensive to QC and convert the entire UK Biobank WGS files to pgens! github.com/chundruv/ukb...
And for All of Us v8, we will provide you with the QC’ed pgen files on publication in a public workspace available to registered users.
And for All of Us v8, we will provide you with the QC’ed pgen files on publication in a public workspace available to registered users.

GitHub - chundruv/ukbb_pvcf2pgen: UKBB pVCF to plink pgen conversion
UKBB pVCF to plink pgen conversion. Contribute to chundruv/ukbb_pvcf2pgen development by creating an account on GitHub.
github.com
November 8, 2025 at 9:31 AM
Using our DNANexus applet it is fast and not too expensive to QC and convert the entire UK Biobank WGS files to pgens! github.com/chundruv/ukb...
And for All of Us v8, we will provide you with the QC’ed pgen files on publication in a public workspace available to registered users.
And for All of Us v8, we will provide you with the QC’ed pgen files on publication in a public workspace available to registered users.
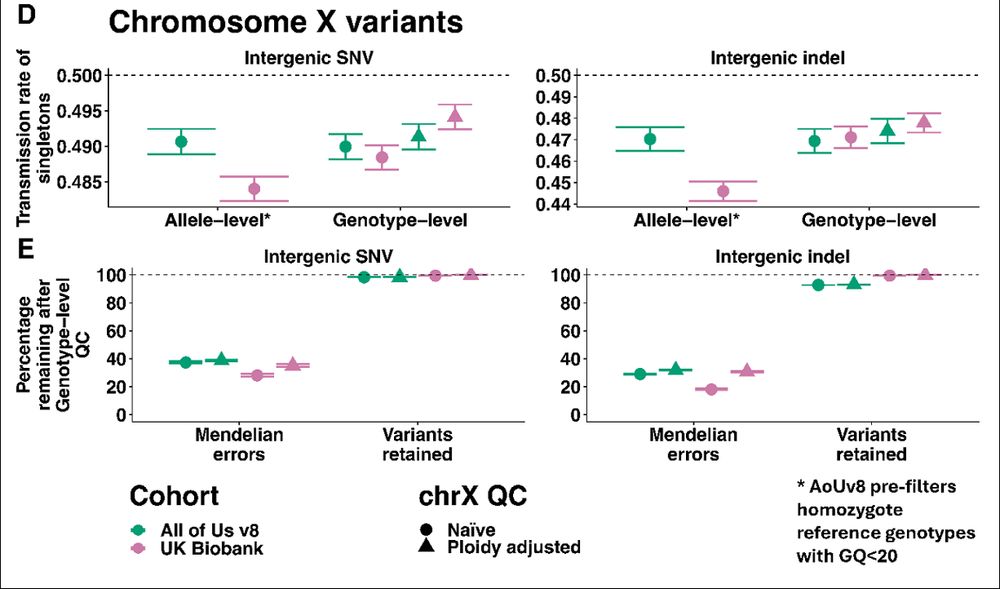
“Ok fine, but what about the X chromosome, you always forget that”
This time we didn’t ignore the X chromosome! We show that you should pay special attention to non-pseudoautosomal X chromosome where QC should be more lenient for haploid males.
This time we didn’t ignore the X chromosome! We show that you should pay special attention to non-pseudoautosomal X chromosome where QC should be more lenient for haploid males.
November 8, 2025 at 9:31 AM
“Ok fine, but what about the X chromosome, you always forget that”
This time we didn’t ignore the X chromosome! We show that you should pay special attention to non-pseudoautosomal X chromosome where QC should be more lenient for haploid males.
This time we didn’t ignore the X chromosome! We show that you should pay special attention to non-pseudoautosomal X chromosome where QC should be more lenient for haploid males.
“But Kartik, how do we know the genotypes are wrong?”
Trios! Both cohorts have ~1k parent-offspring trios that were recruited incidentally.
Applying genotype-level QC reduces Mendelian errors by ~60-80% (even in All of Us where they already did genotype-level QC on hom-refs!)
Trios! Both cohorts have ~1k parent-offspring trios that were recruited incidentally.
Applying genotype-level QC reduces Mendelian errors by ~60-80% (even in All of Us where they already did genotype-level QC on hom-refs!)

November 8, 2025 at 9:31 AM
“But Kartik, how do we know the genotypes are wrong?”
Trios! Both cohorts have ~1k parent-offspring trios that were recruited incidentally.
Applying genotype-level QC reduces Mendelian errors by ~60-80% (even in All of Us where they already did genotype-level QC on hom-refs!)
Trios! Both cohorts have ~1k parent-offspring trios that were recruited incidentally.
Applying genotype-level QC reduces Mendelian errors by ~60-80% (even in All of Us where they already did genotype-level QC on hom-refs!)
“Bah humbug! How bad could it be?”
After genotype-level QC and a 10% missingness cut-off, we remove ~100 million (~9%) variants!
Most genotypes removed are homozygote reference (which were filtered in All of Us already)
After genotype-level QC and a 10% missingness cut-off, we remove ~100 million (~9%) variants!
Most genotypes removed are homozygote reference (which were filtered in All of Us already)

November 8, 2025 at 9:31 AM
“Bah humbug! How bad could it be?”
After genotype-level QC and a 10% missingness cut-off, we remove ~100 million (~9%) variants!
Most genotypes removed are homozygote reference (which were filtered in All of Us already)
After genotype-level QC and a 10% missingness cut-off, we remove ~100 million (~9%) variants!
Most genotypes removed are homozygote reference (which were filtered in All of Us already)
We caution that the released data in UK Biobank and All of Us is not as clean as you may believe!
Here, we show how we determine data quality in WGS data, provide a really fast way of doing so on biobank data, and we will release QC’ed plink files for All of Us upon publication
Here, we show how we determine data quality in WGS data, provide a really fast way of doing so on biobank data, and we will release QC’ed plink files for All of Us upon publication

November 8, 2025 at 9:31 AM
We caution that the released data in UK Biobank and All of Us is not as clean as you may believe!
Here, we show how we determine data quality in WGS data, provide a really fast way of doing so on biobank data, and we will release QC’ed plink files for All of Us upon publication
Here, we show how we determine data quality in WGS data, provide a really fast way of doing so on biobank data, and we will release QC’ed plink files for All of Us upon publication
I missed Hyung Chul’s poster! I’ll have to go see it later today
May 26, 2025 at 8:14 AM
I missed Hyung Chul’s poster! I’ll have to go see it later today