




Kartik Chundru
@chundru.bsky.social
Postdoc at University of Exeter 🇮🇪🇮🇳🇬🇧 Statistical/Computational analyses using any NGS-based data
Formerly at Sanger institute working on recessive developmental disorders in DDD
Formerly at Sanger institute working on recessive developmental disorders in DDD
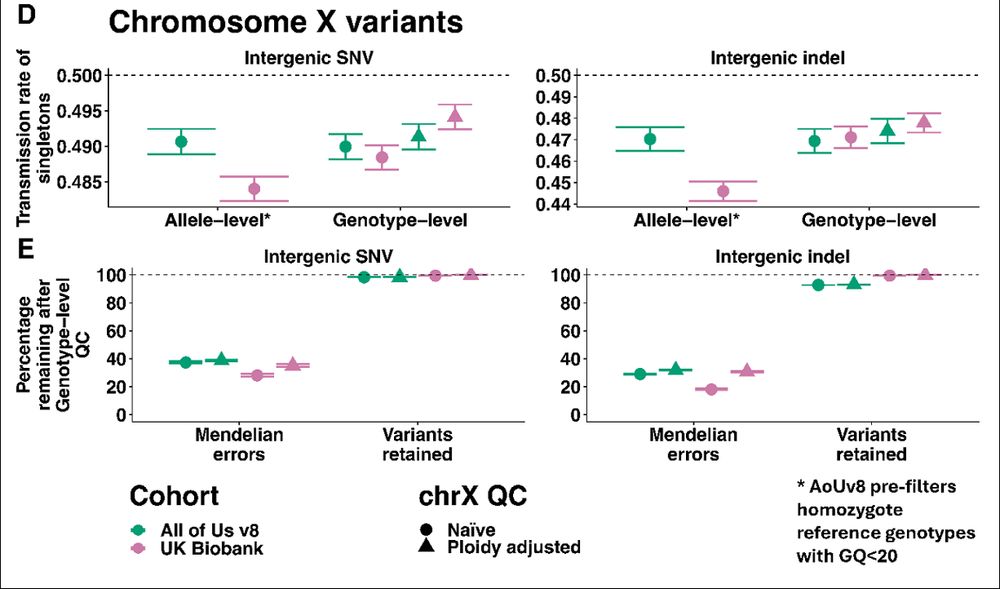
“Ok fine, but what about the X chromosome, you always forget that”
This time we didn’t ignore the X chromosome! We show that you should pay special attention to non-pseudoautosomal X chromosome where QC should be more lenient for haploid males.
This time we didn’t ignore the X chromosome! We show that you should pay special attention to non-pseudoautosomal X chromosome where QC should be more lenient for haploid males.
November 8, 2025 at 9:31 AM
“Ok fine, but what about the X chromosome, you always forget that”
This time we didn’t ignore the X chromosome! We show that you should pay special attention to non-pseudoautosomal X chromosome where QC should be more lenient for haploid males.
This time we didn’t ignore the X chromosome! We show that you should pay special attention to non-pseudoautosomal X chromosome where QC should be more lenient for haploid males.
“But Kartik, how do we know the genotypes are wrong?”
Trios! Both cohorts have ~1k parent-offspring trios that were recruited incidentally.
Applying genotype-level QC reduces Mendelian errors by ~60-80% (even in All of Us where they already did genotype-level QC on hom-refs!)
Trios! Both cohorts have ~1k parent-offspring trios that were recruited incidentally.
Applying genotype-level QC reduces Mendelian errors by ~60-80% (even in All of Us where they already did genotype-level QC on hom-refs!)

November 8, 2025 at 9:31 AM
“But Kartik, how do we know the genotypes are wrong?”
Trios! Both cohorts have ~1k parent-offspring trios that were recruited incidentally.
Applying genotype-level QC reduces Mendelian errors by ~60-80% (even in All of Us where they already did genotype-level QC on hom-refs!)
Trios! Both cohorts have ~1k parent-offspring trios that were recruited incidentally.
Applying genotype-level QC reduces Mendelian errors by ~60-80% (even in All of Us where they already did genotype-level QC on hom-refs!)
“Bah humbug! How bad could it be?”
After genotype-level QC and a 10% missingness cut-off, we remove ~100 million (~9%) variants!
Most genotypes removed are homozygote reference (which were filtered in All of Us already)
After genotype-level QC and a 10% missingness cut-off, we remove ~100 million (~9%) variants!
Most genotypes removed are homozygote reference (which were filtered in All of Us already)

November 8, 2025 at 9:31 AM
“Bah humbug! How bad could it be?”
After genotype-level QC and a 10% missingness cut-off, we remove ~100 million (~9%) variants!
Most genotypes removed are homozygote reference (which were filtered in All of Us already)
After genotype-level QC and a 10% missingness cut-off, we remove ~100 million (~9%) variants!
Most genotypes removed are homozygote reference (which were filtered in All of Us already)
We caution that the released data in UK Biobank and All of Us is not as clean as you may believe!
Here, we show how we determine data quality in WGS data, provide a really fast way of doing so on biobank data, and we will release QC’ed plink files for All of Us upon publication
Here, we show how we determine data quality in WGS data, provide a really fast way of doing so on biobank data, and we will release QC’ed plink files for All of Us upon publication

November 8, 2025 at 9:31 AM
We caution that the released data in UK Biobank and All of Us is not as clean as you may believe!
Here, we show how we determine data quality in WGS data, provide a really fast way of doing so on biobank data, and we will release QC’ed plink files for All of Us upon publication
Here, we show how we determine data quality in WGS data, provide a really fast way of doing so on biobank data, and we will release QC’ed plink files for All of Us upon publication