



Bernhard Bein
@bernhardbein.bsky.social
PhD student in comparative genomics @ Senckenberg and Goethe University Frankfurt
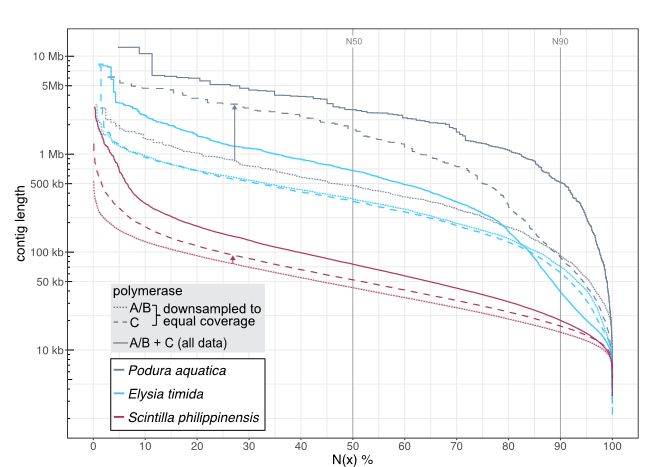
Applying polymerase A/B and C read amplification to two mollusc and one arthropod sample, we saw that assembly contiguity was either higher or comparable with polymerase C reads, and assemblies were always more contigous when combining all libraries, probably due to complementary read drop-outs.
February 10, 2025 at 3:26 PM
Applying polymerase A/B and C read amplification to two mollusc and one arthropod sample, we saw that assembly contiguity was either higher or comparable with polymerase C reads, and assemblies were always more contigous when combining all libraries, probably due to complementary read drop-outs.
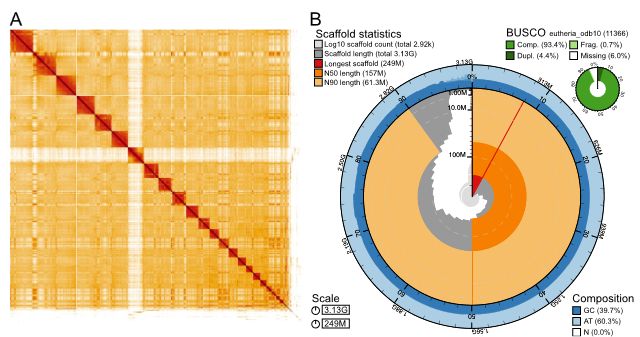
Combining reads of polymerase A/B and C with HiC reads, we were able to assemble the maned sloth genome to chromosome level with a contig N50 of ~4.75 Mb, a 10x increase compared to polymerase A/B reads alone.
The assemblies 3.1 Gb size also surpasses the previous 500 Mb protocol size limit.
The assemblies 3.1 Gb size also surpasses the previous 500 Mb protocol size limit.
February 10, 2025 at 3:26 PM
Combining reads of polymerase A/B and C with HiC reads, we were able to assemble the maned sloth genome to chromosome level with a contig N50 of ~4.75 Mb, a 10x increase compared to polymerase A/B reads alone.
The assemblies 3.1 Gb size also surpasses the previous 500 Mb protocol size limit.
The assemblies 3.1 Gb size also surpasses the previous 500 Mb protocol size limit.
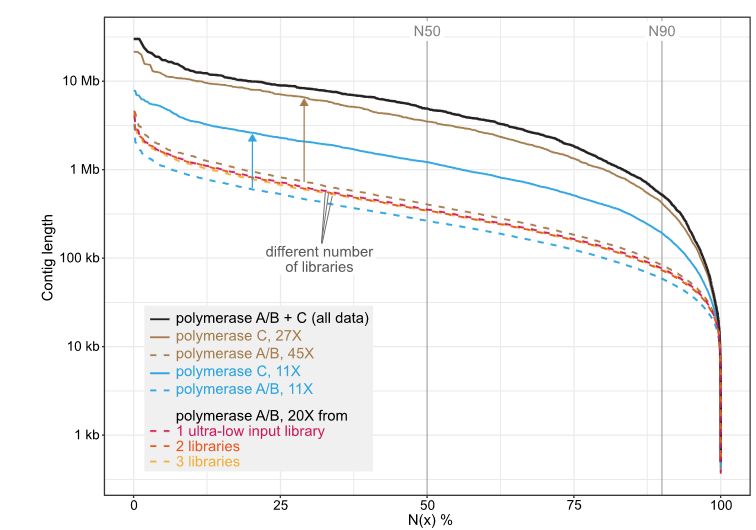
We then used a different polymerase "C" to repeat amplification and sequencing, increasing contig N50 and compleasm/TOGA gene completness dramatically.
Aligning reads of B. torquatus to another sloth species, we saw obvious read drop-outs that polymerase C reads could cover:
Aligning reads of B. torquatus to another sloth species, we saw obvious read drop-outs that polymerase C reads could cover:

February 10, 2025 at 3:26 PM
We then used a different polymerase "C" to repeat amplification and sequencing, increasing contig N50 and compleasm/TOGA gene completness dramatically.
Aligning reads of B. torquatus to another sloth species, we saw obvious read drop-outs that polymerase C reads could cover:
Aligning reads of B. torquatus to another sloth species, we saw obvious read drop-outs that polymerase C reads could cover:
We therefore switched to the ultra-low input protocol, which contains a PCR amplification step of input DNA, employing two different polymerases we call A/B.
This way, we could sequence the maned sloth Bradypus torquatus to a coverage of 45X.
However, contig N50 of the assembly was below 1 Mb:
This way, we could sequence the maned sloth Bradypus torquatus to a coverage of 45X.
However, contig N50 of the assembly was below 1 Mb:
February 10, 2025 at 3:26 PM
We therefore switched to the ultra-low input protocol, which contains a PCR amplification step of input DNA, employing two different polymerases we call A/B.
This way, we could sequence the maned sloth Bradypus torquatus to a coverage of 45X.
However, contig N50 of the assembly was below 1 Mb:
This way, we could sequence the maned sloth Bradypus torquatus to a coverage of 45X.
However, contig N50 of the assembly was below 1 Mb: