




Bernardo Rodríguez Martín
@bernardo-rodriguez.bsky.social
Team Leader & Independent Fellow at the @crg.eu
At the Repetitive DNA Biology (REPBIO) Lab, we leverage the latest technologies to decode nucleotide sequences for investigating how repetitive DNA shapes genome function and contributes to disease.
At the Repetitive DNA Biology (REPBIO) Lab, we leverage the latest technologies to decode nucleotide sequences for investigating how repetitive DNA shapes genome function and contributes to disease.
Reposted by Bernardo Rodríguez Martín

Next was Bernardo Rodriguez-Martín ( @bernardo-rodriguez.bsky.social ), from the EvoMG Program and @crg.eu, on highly mutagenic (hot) L1 polymorphisms in cancer

October 12, 2025 at 9:10 AM
Next was Bernardo Rodriguez-Martín ( @bernardo-rodriguez.bsky.social ), from the EvoMG Program and @crg.eu, on highly mutagenic (hot) L1 polymorphisms in cancer
Thanks to the huge contributions from all the co-authors of this large effort, who are listed in the main thread, and to the reviewers, whose thoughtful feedback served to greatly improve the manuscript since its initial submission 🙏
July 31, 2025 at 7:32 AM
Thanks to the huge contributions from all the co-authors of this large effort, who are listed in the main thread, and to the reviewers, whose thoughtful feedback served to greatly improve the manuscript since its initial submission 🙏
These insights were largely possible thanks to SVAN (github.com/REPBIO-LAB/S...). While many tools call SVs, none systematically analyse their sequences to infer mechanisms. SVAN fills this gap by taking a VCF from your favourite SV caller and adding annotations of SV classes and features [9/9]

GitHub - REPBIO-LAB/SVAN: Structural Variants ANnotator (SVAN)
Structural Variants ANnotator (SVAN). Contribute to REPBIO-LAB/SVAN development by creating an account on GitHub.
github.com
July 31, 2025 at 7:32 AM
These insights were largely possible thanks to SVAN (github.com/REPBIO-LAB/S...). While many tools call SVs, none systematically analyse their sequences to infer mechanisms. SVAN fills this gap by taking a VCF from your favourite SV caller and adding annotations of SV classes and features [9/9]
We identified 10 HERVK, 7 of them not previously reported, and 28 solo-long terminal repeats (LTRs) [7/9]

July 31, 2025 at 7:32 AM
We identified 10 HERVK, 7 of them not previously reported, and 28 solo-long terminal repeats (LTRs) [7/9]
Transduction tracing uncovers locus-specific patterns in source L1 and SVA elements. While most L1s mediate only 3′ transductions, some SVAs show 5′ or 3′ specificity. A striking exception: one highly active L1 uses promoter hijacking to drive 5′ transductions. [6/9]

July 31, 2025 at 7:32 AM
Transduction tracing uncovers locus-specific patterns in source L1 and SVA elements. While most L1s mediate only 3′ transductions, some SVAs show 5′ or 3′ specificity. A striking exception: one highly active L1 uses promoter hijacking to drive 5′ transductions. [6/9]
We observed large sequence diversity among L1 and SVA inserts, with dozens of structural conformations shaped by their integration mechanisms and transductions. SVA elements show a broad size range, largely driven by variability in their internal hexameric and VNTR repeats. [5/9]

July 31, 2025 at 7:32 AM
We observed large sequence diversity among L1 and SVA inserts, with dozens of structural conformations shaped by their integration mechanisms and transductions. SVA elements show a broad size range, largely driven by variability in their internal hexameric and VNTR repeats. [5/9]
Our new study scales up to the population level: we sequenced 1,019 human genomes with ONT, resolving the sequence of 23,212 Alu, 4,851 L1, and 3,239 SVA insertions, a gain of +20% (Alu), +166% (L1), and +179% (SVA) over short-read analysis of the same samples. [4/9]

July 31, 2025 at 7:32 AM
Our new study scales up to the population level: we sequenced 1,019 human genomes with ONT, resolving the sequence of 23,212 Alu, 4,851 L1, and 3,239 SVA insertions, a gain of +20% (Alu), +166% (L1), and +179% (SVA) over short-read analysis of the same samples. [4/9]
Refreshment of Chapter 2: “Twin priming”.
Microhomology patterns observed at 5´ inversion and truncation breakpoint junctions for 1.271 L1 insertions point to MMEJ. We also found three templated insertions, suggestive of polymerase theta. [3/9]
www.cell.com/cell/fulltex...
Microhomology patterns observed at 5´ inversion and truncation breakpoint junctions for 1.271 L1 insertions point to MMEJ. We also found three templated insertions, suggestive of polymerase theta. [3/9]
www.cell.com/cell/fulltex...

Recurrent inversion polymorphisms in humans associate with genetic instability and genomic disorders
Large-scale analysis of haplotype-resolved inversions in human genomes unveils recurrent
inversion polymorphisms and their disease relevance.
www.cell.com
July 31, 2025 at 7:32 AM
Refreshment of Chapter 2: “Twin priming”.
Microhomology patterns observed at 5´ inversion and truncation breakpoint junctions for 1.271 L1 insertions point to MMEJ. We also found three templated insertions, suggestive of polymerase theta. [3/9]
www.cell.com/cell/fulltex...
Microhomology patterns observed at 5´ inversion and truncation breakpoint junctions for 1.271 L1 insertions point to MMEJ. We also found three templated insertions, suggestive of polymerase theta. [3/9]
www.cell.com/cell/fulltex...
Refreshment of Chapter 1: “Hot L1s, age and coding potential”.
MEI analysis on 32 haplotype-resolved human genomes. Older L1s have higher allele frequencies, lower ORF preservation and are therefore less active, but eternal youth can occur [2/9]
www.science.org/doi/10.1126/...
MEI analysis on 32 haplotype-resolved human genomes. Older L1s have higher allele frequencies, lower ORF preservation and are therefore less active, but eternal youth can occur [2/9]
www.science.org/doi/10.1126/...

Haplotype-resolved diverse human genomes and integrated analysis of structural variation
Human genetic variation is elucidated from de novo assembly of 32 genomes selected as representatives of the 1000 Genomes Project.
www.science.org
July 31, 2025 at 7:32 AM
Refreshment of Chapter 1: “Hot L1s, age and coding potential”.
MEI analysis on 32 haplotype-resolved human genomes. Older L1s have higher allele frequencies, lower ORF preservation and are therefore less active, but eternal youth can occur [2/9]
www.science.org/doi/10.1126/...
MEI analysis on 32 haplotype-resolved human genomes. Older L1s have higher allele frequencies, lower ORF preservation and are therefore less active, but eternal youth can occur [2/9]
www.science.org/doi/10.1126/...
Reposted by Bernardo Rodríguez Martín

The first, co-authored by @bernardo-rodriguez.bsky.social, discovered more than 167,000 structural variants across the 1,019 individuals, doubling the known amount of structural variation in the human pangenome. Most variants were rare, which will help accelerate the diagnosis of rare diseases.
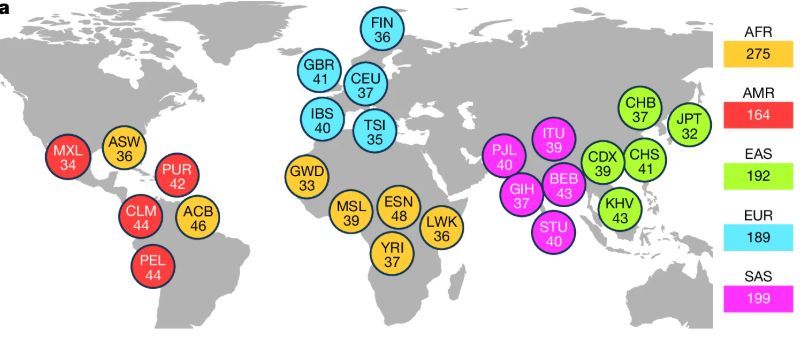
July 23, 2025 at 3:50 PM
The first, co-authored by @bernardo-rodriguez.bsky.social, discovered more than 167,000 structural variants across the 1,019 individuals, doubling the known amount of structural variation in the human pangenome. Most variants were rare, which will help accelerate the diagnosis of rare diseases.