



Sergey Nurk
@sergeynurk.bsky.social
Principal Bionformatician
@nanopore. Ex: Postdoctoral fellow @ NIH; Researcher @ CAB. Views are my own; #StandWithUkraine Support Ukraine!
@nanopore. Ex: Postdoctoral fellow @ NIH; Researcher @ CAB. Views are my own; #StandWithUkraine Support Ukraine!
Reposted by Sergey Nurk

Fantastic talk by @vikramshivakumar.bsky.social Mumemto—Scalable multi-MUM finding for pangenomes
Papers biorxiv.org/content/10.1101/2025.05.20.654611 & doi.org/10.1186/s13059-025-03644-0
Code: github.com/vikshiv/mume...
Very efficient pangenome visualization tool, revealing synteny and variations!
Papers biorxiv.org/content/10.1101/2025.05.20.654611 & doi.org/10.1186/s13059-025-03644-0
Code: github.com/vikshiv/mume...
Very efficient pangenome visualization tool, revealing synteny and variations!



November 6, 2025 at 1:13 AM
Fantastic talk by @vikramshivakumar.bsky.social Mumemto—Scalable multi-MUM finding for pangenomes
Papers biorxiv.org/content/10.1101/2025.05.20.654611 & doi.org/10.1186/s13059-025-03644-0
Code: github.com/vikshiv/mume...
Very efficient pangenome visualization tool, revealing synteny and variations!
Papers biorxiv.org/content/10.1101/2025.05.20.654611 & doi.org/10.1186/s13059-025-03644-0
Code: github.com/vikshiv/mume...
Very efficient pangenome visualization tool, revealing synteny and variations!
Reposted by Sergey Nurk

Thread on #GI2025 's second day! 👇🏻
Second day of Genome Informatics #GI2025 began with the session “Genome Assembly and Sequence Algorithms" Yun William Yu presented “Average-case Analysis of Seed-Chain-Extend under Random Mutations"
genome.cshlp.org/content/33/7/1175
providing theoretical guarantees for the popular seed-chain-extend
genome.cshlp.org/content/33/7/1175
providing theoretical guarantees for the popular seed-chain-extend


November 6, 2025 at 5:53 PM
Thread on #GI2025 's second day! 👇🏻
Reposted by Sergey Nurk

Reposted by Sergey Nurk

Following ish's `filter` and bqtools' `grep`, Sassy now also has initial support for grep and filter!
Grep mode shows all matches, grouped per record, and is meant for human consumption.
Filter mode prints full matching (or non-matching) records to stdout or output files.
Grep mode shows all matches, grouped per record, and is meant for human consumption.
Filter mode prints full matching (or non-matching) records to stdout or output files.

October 30, 2025 at 11:46 PM
Following ish's `filter` and bqtools' `grep`, Sassy now also has initial support for grep and filter!
Grep mode shows all matches, grouped per record, and is meant for human consumption.
Filter mode prints full matching (or non-matching) records to stdout or output files.
Grep mode shows all matches, grouped per record, and is meant for human consumption.
Filter mode prints full matching (or non-matching) records to stdout or output files.
Reposted by Sergey Nurk

Very excited about Movi 2! Excellent work by Mohsen here. FYI, I have a series of 5 videos on the move structure starting with this one: youtu.be/REniD2dKf6A?...
October 21, 2025 at 9:39 PM
Very excited about Movi 2! Excellent work by Mohsen here. FYI, I have a series of 5 videos on the move structure starting with this one: youtu.be/REniD2dKf6A?...
Reposted by Sergey Nurk

ASHG Plenary Session starting with the awards ceremony honoring Eric Green with the Leadership Award of @geneticssociety.bsky.social reflecting on his career in human genetics & genomics leading the Human Genome Project & the NHGRI and the leadership principles he has learned throughout #ASHG25


October 16, 2025 at 8:47 PM
ASHG Plenary Session starting with the awards ceremony honoring Eric Green with the Leadership Award of @geneticssociety.bsky.social reflecting on his career in human genetics & genomics leading the Human Genome Project & the NHGRI and the leadership principles he has learned throughout #ASHG25
Reposted by Sergey Nurk

The T2T zebra finch genome has hatched! 🐣 🧬 @vertebrategenomes.bsky.social

The complete genome of a songbird https://www.biorxiv.org/content/10.1101/2025.10.14.682431v1
October 15, 2025 at 1:08 PM
The T2T zebra finch genome has hatched! 🐣 🧬 @vertebrategenomes.bsky.social
Reposted by Sergey Nurk

I am hiring! - looking for a Staff Scientist to co-run my research group with me. Staff Scientist is a senior professional scientist role at EMBL. Please forward to people you might know who could be interested! embl.wd103.myworkdayjobs.com/en-US/EMBL/j...

Staff Scientist
About EMBL-EBI EMBL’s European Bioinformatics Institute is a data powerhouse, utilised on a global scale to advance scientific discovery through bioinformatics and solutions to some of the world’s mos...
embl.wd103.myworkdayjobs.com
October 10, 2025 at 7:30 AM
I am hiring! - looking for a Staff Scientist to co-run my research group with me. Staff Scientist is a senior professional scientist role at EMBL. Please forward to people you might know who could be interested! embl.wd103.myworkdayjobs.com/en-US/EMBL/j...
Reposted by Sergey Nurk

The Metagraph paper is out in Nature; it showed up in my feeds today! Congratulations to Mikhail Karasikov, @gxxxr.bsky.social, @akkah21.bsky.social and all of the other authors (whom I'd love to follow on Bluesky if I can find you ;P) www.nature.com/articles/s41...

Efficient and accurate search in petabase-scale sequence repositories - Nature
MetaGraph enables scalable indexing of large sets of DNA, RNA or protein sequences using annotated de Bruijn graphs.
www.nature.com
October 9, 2025 at 2:40 PM
The Metagraph paper is out in Nature; it showed up in my feeds today! Congratulations to Mikhail Karasikov, @gxxxr.bsky.social, @akkah21.bsky.social and all of the other authors (whom I'd love to follow on Bluesky if I can find you ;P) www.nature.com/articles/s41...
Reposted by Sergey Nurk
I've added 7 videos to my Burrows-Wheeler indexing playlist (www.youtube.com/playlist?lis...), rounding out the r-index series and adding a 5-part series on the move structure. Now 27 videos in that playlist. I aim to add videos on prefix-free parsing, PBWT, Wheeler languages/automata in the future.

Burrows-Wheeler Indexing - YouTube
Videos on : (a) the Burrows-Wheeler Transform (BWT), (b) the FM Index, which uses the BWT to construct a full-text index, (c) Wheeler graphs, (d) r-index, an...
www.youtube.com
October 7, 2025 at 2:17 PM
I've added 7 videos to my Burrows-Wheeler indexing playlist (www.youtube.com/playlist?lis...), rounding out the r-index series and adding a 5-part series on the move structure. Now 27 videos in that playlist. I aim to add videos on prefix-free parsing, PBWT, Wheeler languages/automata in the future.
Reposted by Sergey Nurk

🦒Long read giraffe is out!🦒
Mapping long reads to pangenome graphs is ~10x faster than with GraphAligner, with veeery slightly better mapping accuracy, short variant calling, and SV genotyping than GraphAligner or Minimap2
Mapping long reads to pangenome graphs is ~10x faster than with GraphAligner, with veeery slightly better mapping accuracy, short variant calling, and SV genotyping than GraphAligner or Minimap2
Rapid, accurate long- and short-read mapping to large pangenome graphs with vg Giraffe https://www.biorxiv.org/content/10.1101/2025.09.29.678807v1
October 2, 2025 at 6:28 AM
🦒Long read giraffe is out!🦒
Mapping long reads to pangenome graphs is ~10x faster than with GraphAligner, with veeery slightly better mapping accuracy, short variant calling, and SV genotyping than GraphAligner or Minimap2
Mapping long reads to pangenome graphs is ~10x faster than with GraphAligner, with veeery slightly better mapping accuracy, short variant calling, and SV genotyping than GraphAligner or Minimap2
Reposted by Sergey Nurk
Do you know ~60% of human SVs fall in ~1% of GRCh38? See our new preprint: arxiv.org/abs/2509.23057 and the companion blog post on how we started this project and longdust: lh3.github.io/2025/09/29/o.... Work with Alvin Qin

September 30, 2025 at 2:19 AM
Do you know ~60% of human SVs fall in ~1% of GRCh38? See our new preprint: arxiv.org/abs/2509.23057 and the companion blog post on how we started this project and longdust: lh3.github.io/2025/09/29/o.... Work with Alvin Qin
Reposted by Sergey Nurk

Delighted to see our paper studying the evolution of plasmids over the last 100 years, now out! Years of work by Adrian Cazares, also Nick Thomson @sangerinstitute.bsky.social - this version much improved over the preprint. Final version should be open access, apols.
Thread 1/n
Thread 1/n

September 25, 2025 at 9:29 PM
Delighted to see our paper studying the evolution of plasmids over the last 100 years, now out! Years of work by Adrian Cazares, also Nick Thomson @sangerinstitute.bsky.social - this version much improved over the preprint. Final version should be open access, apols.
Thread 1/n
Thread 1/n
Reposted by Sergey Nurk
Delighted to finally announce a preprint describing the Q100 project! “A complete diploid human genome benchmark for personalized genomics” For which we finished HG002 to near-perfect accuracy: www.biorxiv.org/content/10.1... 🧵[1/14]

A complete diploid human genome benchmark for personalized genomics
Human genome resequencing typically involves mapping reads to a reference genome to call variants; however, this approach suffers from both technical and reference biases, leaving many duplicated and ...
www.biorxiv.org
September 22, 2025 at 5:01 PM
Delighted to finally announce a preprint describing the Q100 project! “A complete diploid human genome benchmark for personalized genomics” For which we finished HG002 to near-perfect accuracy: www.biorxiv.org/content/10.1... 🧵[1/14]
Reposted by Sergey Nurk
Excited to share our EvANI benchmarking workflow, published in Briefings in Bioinformatics doi.org/10.1093/bib/...
Computing average nucleotide identity (ANI) is neither conceptually nor computationally trivial. Its definition has evolved over years, with different meanings and assumptions (1/5)
Computing average nucleotide identity (ANI) is neither conceptually nor computationally trivial. Its definition has evolved over years, with different meanings and assumptions (1/5)

September 21, 2025 at 3:26 PM
Excited to share our EvANI benchmarking workflow, published in Briefings in Bioinformatics doi.org/10.1093/bib/...
Computing average nucleotide identity (ANI) is neither conceptually nor computationally trivial. Its definition has evolved over years, with different meanings and assumptions (1/5)
Computing average nucleotide identity (ANI) is neither conceptually nor computationally trivial. Its definition has evolved over years, with different meanings and assumptions (1/5)
Reposted by Sergey Nurk

MMseqs2-GPU sets new standards in single query search speed, allows near instant search of big databases, scales to multiple GPUs and is fast beyond VRAM. It enables ColabFold MSA generation in seconds and sub-second Foldseek search against AFDB50. 1/n
📄 www.nature.com/articles/s41...
💿 mmseqs.com
📄 www.nature.com/articles/s41...
💿 mmseqs.com

GPU-accelerated homology search with MMseqs2 - Nature Methods
Graphics processing unit-accelerated MMseqs2 offers tremendous speedups for homology retrieval from metagenomic databases, query-centered multiple sequence alignment generation for structure predictio...
www.nature.com
September 21, 2025 at 8:06 AM
MMseqs2-GPU sets new standards in single query search speed, allows near instant search of big databases, scales to multiple GPUs and is fast beyond VRAM. It enables ColabFold MSA generation in seconds and sub-second Foldseek search against AFDB50. 1/n
📄 www.nature.com/articles/s41...
💿 mmseqs.com
📄 www.nature.com/articles/s41...
💿 mmseqs.com
Reposted by Sergey Nurk

Some very interesting preliminary benchmarks between Stringzilla and some Rust libs from Ash: x.com/ashvardanian...
Including some alignment comparisons.
Including some alignment comparisons.

Ash Vardanian on X: "Let there be https://t.co/KbIf9uMiVC 🧵 So many great libraries in Rust - the perfect soil for a thematic benchmark - across hashing, similarity scoring, search, and sketching Write-up coming, but there are already some indicative intermediate results https://t.co/B4rGLllnXM" / X
Let there be https://t.co/KbIf9uMiVC 🧵 So many great libraries in Rust - the perfect soil for a thematic benchmark - across hashing, similarity scoring, search, and sketching Write-up coming, but there are already some indicative intermediate results https://t.co/B4rGLllnXM
x.com
September 12, 2025 at 12:21 AM
Some very interesting preliminary benchmarks between Stringzilla and some Rust libs from Ash: x.com/ashvardanian...
Including some alignment comparisons.
Including some alignment comparisons.
Reposted by Sergey Nurk
Now preprinted at arxiv.org/abs/2509.07357

September 10, 2025 at 2:10 AM
Now preprinted at arxiv.org/abs/2509.07357
Reposted by Sergey Nurk

Preprint out for myloasm, our new nanopore / HiFi metagenome assembler!
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
High-resolution metagenome assembly for modern long reads with myloasm https://www.biorxiv.org/content/10.1101/2025.09.05.674543v1
September 7, 2025 at 11:35 PM
Preprint out for myloasm, our new nanopore / HiFi metagenome assembler!
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
Reposted by Sergey Nurk
For anyone who has used pling for comparing plasmids using rearrangement distances ("how many structural events apart are these plasmids"), here's how to tweak parameters, and integrate it with typing info, and the host phylogeny
www.biorxiv.org/content/10.1...
github.com/iqbal-lab-or...
www.biorxiv.org/content/10.1...
github.com/iqbal-lab-or...

Clustering of plasmid genomes for genomic epidemiology by using rearrangement distances, with pling
Integration of plasmids into genomic epidemiology is challenging, because there are no clearly defined evolving-units (equivalent to species), and because plasmids appear to evolve as much by structur...
www.biorxiv.org
September 7, 2025 at 2:56 PM
For anyone who has used pling for comparing plasmids using rearrangement distances ("how many structural events apart are these plasmids"), here's how to tweak parameters, and integrate it with typing info, and the host phylogeny
www.biorxiv.org/content/10.1...
github.com/iqbal-lab-or...
www.biorxiv.org/content/10.1...
github.com/iqbal-lab-or...
Reposted by Sergey Nurk

High-resolution metagenome assembly for modern long reads with myloasm https://www.biorxiv.org/content/10.1101/2025.09.05.674543v1
September 7, 2025 at 4:47 AM
High-resolution metagenome assembly for modern long reads with myloasm https://www.biorxiv.org/content/10.1101/2025.09.05.674543v1
Reposted by Sergey Nurk

🌎👩🔬 For 15+ years biology has accumulated petabytes (million gigabytes) of🧬DNA sequencing data🧬 from the far reaches of our planet.🦠🍄🌵
Logan now democratizes efficient access to the world’s most comprehensive genetics dataset. Free and open.
doi.org/10.1101/2024...
Logan now democratizes efficient access to the world’s most comprehensive genetics dataset. Free and open.
doi.org/10.1101/2024...

September 3, 2025 at 8:39 AM
🌎👩🔬 For 15+ years biology has accumulated petabytes (million gigabytes) of🧬DNA sequencing data🧬 from the far reaches of our planet.🦠🍄🌵
Logan now democratizes efficient access to the world’s most comprehensive genetics dataset. Free and open.
doi.org/10.1101/2024...
Logan now democratizes efficient access to the world’s most comprehensive genetics dataset. Free and open.
doi.org/10.1101/2024...
Reposted by Sergey Nurk

We are glad to announce that the next workshop “Data Structures in Bioinformatics” (DSB 2026) will take place in Venice, Italy, on *February 18-19*, 2026. dsb-meeting.github.io/DSB2026/ Book the dates! #DSB26
DSB 2026 Venice - February 18-19
Workshop Data Structures in Bioinformatics
dsb-meeting.github.io
September 1, 2025 at 6:10 PM
We are glad to announce that the next workshop “Data Structures in Bioinformatics” (DSB 2026) will take place in Venice, Italy, on *February 18-19*, 2026. dsb-meeting.github.io/DSB2026/ Book the dates! #DSB26
Reposted by Sergey Nurk
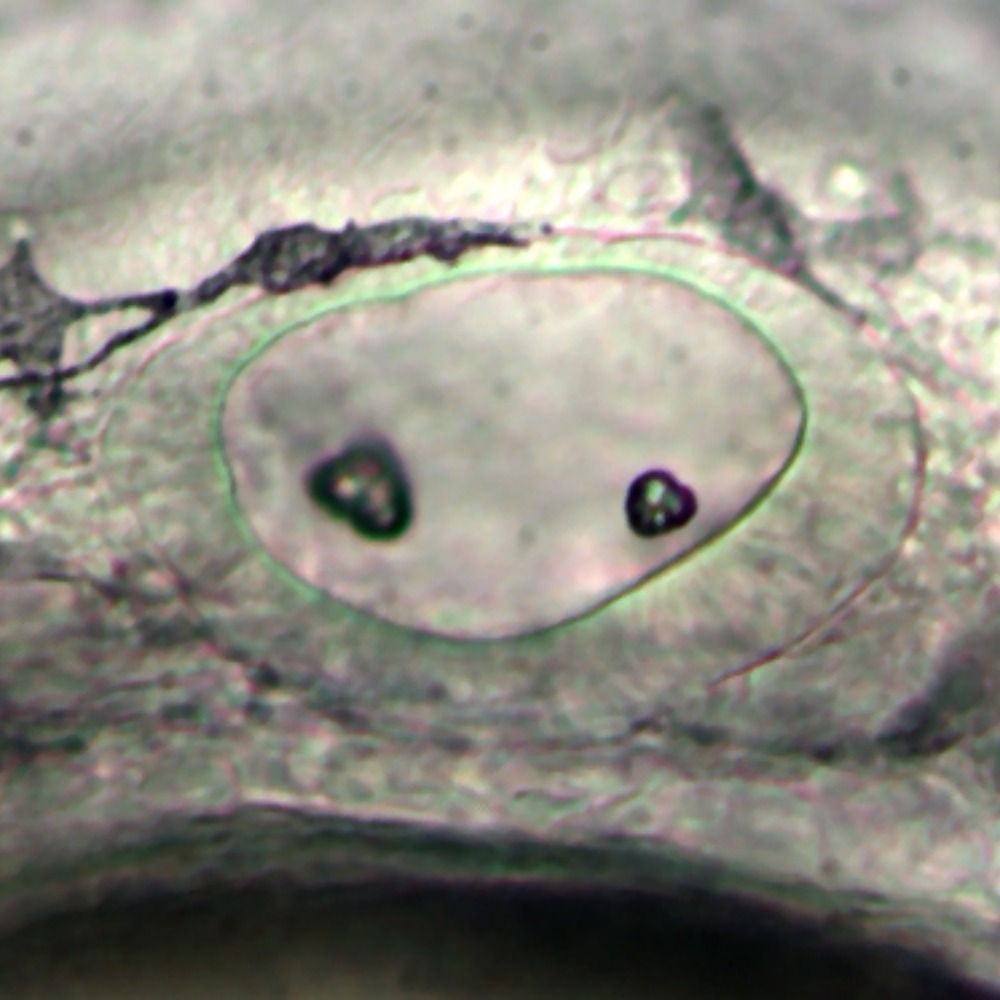
#zebrafish genome update, our T2T assembly of the inbred strain of AB (M-AB) generated by my buddy Nori Sakai has now been released at NCBI and will be a second reference genome for zebrafish (GRCz12ab):
JBQAYU000000000.1 Danio rerio :: NCBI
www.ncbi.nlm.nih.gov
August 15, 2025 at 4:17 PM
#zebrafish genome update, our T2T assembly of the inbred strain of AB (M-AB) generated by my buddy Nori Sakai has now been released at NCBI and will be a second reference genome for zebrafish (GRCz12ab):
Reposted by Sergey Nurk

Excited to share work with
Zhidian Zhang, @milot.bsky.social, @martinsteinegger.bsky.social, and @sokrypton.org
biorxiv.org/content/10.1...
TLDR: We introduce MSA Pairformer, a 111M parameter protein language model that challenges the scaling paradigm in self-supervised protein language modeling🧵
Zhidian Zhang, @milot.bsky.social, @martinsteinegger.bsky.social, and @sokrypton.org
biorxiv.org/content/10.1...
TLDR: We introduce MSA Pairformer, a 111M parameter protein language model that challenges the scaling paradigm in self-supervised protein language modeling🧵

Scaling down protein language modeling with MSA Pairformer
Recent efforts in protein language modeling have focused on scaling single-sequence models and their training data, requiring vast compute resources that limit accessibility. Although models that use ...
biorxiv.org
August 5, 2025 at 6:31 AM
Excited to share work with
Zhidian Zhang, @milot.bsky.social, @martinsteinegger.bsky.social, and @sokrypton.org
biorxiv.org/content/10.1...
TLDR: We introduce MSA Pairformer, a 111M parameter protein language model that challenges the scaling paradigm in self-supervised protein language modeling🧵
Zhidian Zhang, @milot.bsky.social, @martinsteinegger.bsky.social, and @sokrypton.org
biorxiv.org/content/10.1...
TLDR: We introduce MSA Pairformer, a 111M parameter protein language model that challenges the scaling paradigm in self-supervised protein language modeling🧵