



Gonzalo Benegas
@gonzalobenegas.bsky.social
Comp Bio Postdoc @ UC Berkeley
https://gonzalobenegas.github.io/
https://gonzalobenegas.github.io/
Reposted by Gonzalo Benegas

We are excited to share GPN-Star, a cost-effective, biologically grounded genomic language modeling framework that achieves state-of-the-art performance across a wide range of variant effect prediction tasks relevant to human genetics.
www.biorxiv.org/content/10.1...
(1/n)
www.biorxiv.org/content/10.1...
(1/n)

September 22, 2025 at 5:29 AM
We are excited to share GPN-Star, a cost-effective, biologically grounded genomic language modeling framework that achieves state-of-the-art performance across a wide range of variant effect prediction tasks relevant to human genetics.
www.biorxiv.org/content/10.1...
(1/n)
www.biorxiv.org/content/10.1...
(1/n)
Reposted by Gonzalo Benegas

I am thrilled to announce that in January 2026 I will be starting my own lab at NYU Biology! Soon enough I will be recruiting postdocs and students! Please reach out if you are interested with a CV and description of your research interests, or if you know of people who could be interested! 🧬🗽 🦊
June 25, 2025 at 8:10 PM
I am thrilled to announce that in January 2026 I will be starting my own lab at NYU Biology! Soon enough I will be recruiting postdocs and students! Please reach out if you are interested with a CV and description of your research interests, or if you know of people who could be interested! 🧬🗽 🦊
Reposted by Gonzalo Benegas
How can one efficiently simulate phylodynamics for populations with billions of individuals, as is typical in many applications, e.g., viral evolution and cancer genomics? In this work with M. Celentano, @wsdewitt.github.io , & S. Prillo, we provide a solution. doi.org/10.1073/pnas...
1/n
1/n

May 23, 2025 at 9:02 PM
How can one efficiently simulate phylodynamics for populations with billions of individuals, as is typical in many applications, e.g., viral evolution and cancer genomics? In this work with M. Celentano, @wsdewitt.github.io , & S. Prillo, we provide a solution. doi.org/10.1073/pnas...
1/n
1/n
Reposted by Gonzalo Benegas
Thrilled to see my digital art on the cover of Trends Genet. The two binary strings represent reverse-complementary DNA sequences (00=A, 01=C, 10=G, 11=T) and the connecting rectangles represent “embeddings” learned by DNA language models. Pls check out our article as well: doi.org/10.1016/j.ti...

April 7, 2025 at 3:01 PM
Thrilled to see my digital art on the cover of Trends Genet. The two binary strings represent reverse-complementary DNA sequences (00=A, 01=C, 10=G, 11=T) and the connecting rectangles represent “embeddings” learned by DNA language models. Pls check out our article as well: doi.org/10.1016/j.ti...
Reposted by Gonzalo Benegas
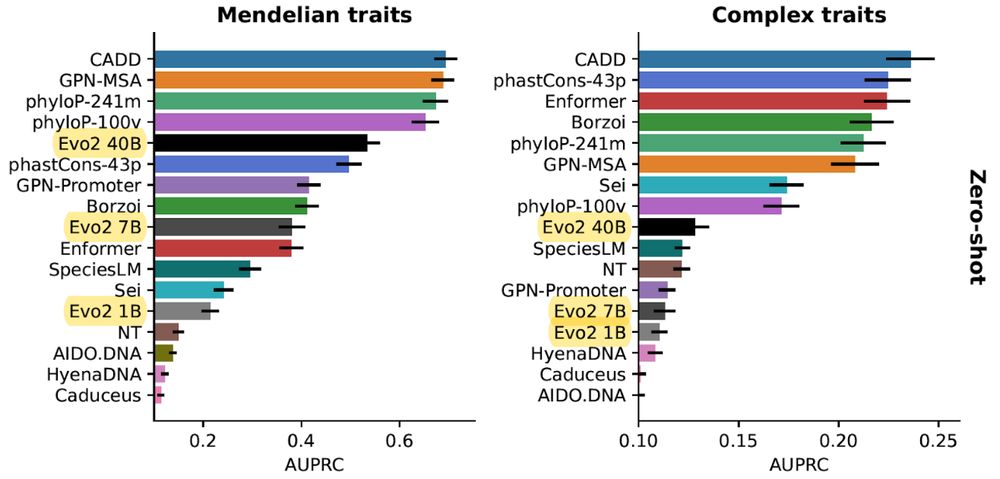
In our updated TraitGym preprint (w/ @gonzalobenegas.bsky.social & Gökcen Eraslan), we evaluate Evo 2 on regulatory variants associated with human traits. We see marked performance gains with scale on Mendelian traits, although still a bit behind alignment-based methods.
doi.org/10.1101/2025...
1/n
doi.org/10.1101/2025...
1/n
March 4, 2025 at 7:54 PM
In our updated TraitGym preprint (w/ @gonzalobenegas.bsky.social & Gökcen Eraslan), we evaluate Evo 2 on regulatory variants associated with human traits. We see marked performance gains with scale on Mendelian traits, although still a bit behind alignment-based methods.
doi.org/10.1101/2025...
1/n
doi.org/10.1101/2025...
1/n
Can DNA sequence models predict mutations affecting human traits?
We introduce TraitGym, a curated benchmark of causal regulatory variants for 113 Mendelian & 83 complex traits, and evaluate functional genomics and DNA language models. Joint work w/ Gökcen Eraslan and @yun-s-song.bsky.social 🧵👇
We introduce TraitGym, a curated benchmark of causal regulatory variants for 113 Mendelian & 83 complex traits, and evaluate functional genomics and DNA language models. Joint work w/ Gökcen Eraslan and @yun-s-song.bsky.social 🧵👇

February 13, 2025 at 8:57 PM
Can DNA sequence models predict mutations affecting human traits?
We introduce TraitGym, a curated benchmark of causal regulatory variants for 113 Mendelian & 83 complex traits, and evaluate functional genomics and DNA language models. Joint work w/ Gökcen Eraslan and @yun-s-song.bsky.social 🧵👇
We introduce TraitGym, a curated benchmark of causal regulatory variants for 113 Mendelian & 83 complex traits, and evaluate functional genomics and DNA language models. Joint work w/ Gökcen Eraslan and @yun-s-song.bsky.social 🧵👇
Reposted by Gonzalo Benegas

Benchmarking DNA Sequence Models for Causal Regulatory Variant Prediction in Human Genetics https://www.biorxiv.org/content/10.1101/2025.02.11.637758v1
February 13, 2025 at 7:33 AM
Benchmarking DNA Sequence Models for Causal Regulatory Variant Prediction in Human Genetics https://www.biorxiv.org/content/10.1101/2025.02.11.637758v1
Reposted by Gonzalo Benegas
Our work, which shows statistical issues with the previous claim of a severe ancient bottleneck in the ancestry of African populations, has been selected as a Featured article in Genetics.
doi.org/10.1093/gene...
doi.org/10.1093/gene...

A previously reported bottleneck in human ancestry 900 kya is likely a statistical artifact
Hu et al. (Science, 2023) recently inferred a severe ancient bottleneck around 900 thousand years (kya) ago in African ancestry but found no similar eviden
doi.org
January 8, 2025 at 8:23 PM
Our work, which shows statistical issues with the previous claim of a severe ancient bottleneck in the ancestry of African populations, has been selected as a Featured article in Genetics.
doi.org/10.1093/gene...
doi.org/10.1093/gene...
Reposted by Gonzalo Benegas
Coincidentally, another article from my lab on DNA language models got published on the same day as GPN-MSA. It's freely available for 50 days from this link:
authors.elsevier.com/a/1kNCscQbJB...
Genomic language models: opportunities and challenges
Please share with your colleagues.
authors.elsevier.com/a/1kNCscQbJB...
Genomic language models: opportunities and challenges
Please share with your colleagues.
authors.elsevier.com
January 3, 2025 at 2:29 AM
Coincidentally, another article from my lab on DNA language models got published on the same day as GPN-MSA. It's freely available for 50 days from this link:
authors.elsevier.com/a/1kNCscQbJB...
Genomic language models: opportunities and challenges
Please share with your colleagues.
authors.elsevier.com/a/1kNCscQbJB...
Genomic language models: opportunities and challenges
Please share with your colleagues.
Reposted by Gonzalo Benegas
Happy New Year! Our GPN-MSA paper is finally published, under a slightly different title from the preprint. Please check it out and share it with your colleagues:
doi.org/10.1038/s415...
1/4
doi.org/10.1038/s415...
1/4

A DNA language model based on multispecies alignment predicts the effects of genome-wide variants - Nature Biotechnology
A language model predicts the effects of genetic variants in the human genome.
doi.org
January 2, 2025 at 8:24 PM
Happy New Year! Our GPN-MSA paper is finally published, under a slightly different title from the preprint. Please check it out and share it with your colleagues:
doi.org/10.1038/s415...
1/4
doi.org/10.1038/s415...
1/4
Reposted by Gonzalo Benegas

A DNA language model based on multispecies alignment predicts the effects of genome-wide variants - @yun-s-song.bsky.social go.nature.com/4gWppWg

A DNA language model based on multispecies alignment predicts the effects of genome-wide variants - Nature Biotechnology
A language model predicts the effects of genetic variants in the human genome.
go.nature.com
January 2, 2025 at 4:18 PM
A DNA language model based on multispecies alignment predicts the effects of genome-wide variants - @yun-s-song.bsky.social go.nature.com/4gWppWg
Reposted by Gonzalo Benegas
Large protein language models can learn complex epistatic interactions, but how much does that help with predicting variant effects? In this NeurIPS article, we show that classical independent-sites phylogenetic models can outperform pLMs on this task.
1/7
openreview.net/forum?id=H7m...
1/7
openreview.net/forum?id=H7m...

Ultrafast classical phylogenetic method beats large protein...
Amino acid substitution rate matrices are fundamental to statistical phylogenetics and evolutionary biology. Estimating them typically requires reconstructed trees for massive amounts of aligned...
openreview.net
November 16, 2024 at 8:42 PM
Large protein language models can learn complex epistatic interactions, but how much does that help with predicting variant effects? In this NeurIPS article, we show that classical independent-sites phylogenetic models can outperform pLMs on this task.
1/7
openreview.net/forum?id=H7m...
1/7
openreview.net/forum?id=H7m...