



Sumit Mukherjee
@smukherjee89.bsky.social
Senior Manager @ Oracle Health. Interested in clinical LLMs, ML + genetics!! Ex-insitro, Microsoft AI4Good, UW. Biriyani expert. Kolkata-> Seattle. Views are my own.
Scholar: https://shorturl.at/QTBGR
Linkedin: https://shorturl.at/WuGl
Scholar: https://shorturl.at/QTBGR
Linkedin: https://shorturl.at/WuGl
(b) Another approach is to generate a more generalized unbiased estimator (see below) that leverages effect estimates from: i) true phenotypes, ii) predicted phenotypes on the same set of sampels, iii) predicted phenotypes on a different set of samples. (8/)

December 23, 2024 at 2:35 AM
(b) Another approach is to generate a more generalized unbiased estimator (see below) that leverages effect estimates from: i) true phenotypes, ii) predicted phenotypes on the same set of sampels, iii) predicted phenotypes on a different set of samples. (8/)
So what is the solution? There are a few different approaches I have seen. (a) you can make parametric assumptions about the relationship between the predicted and true phenotype, which can then be used to 'correct the effect estimates' as in this paper: www.biorxiv.org/content/10.1.... (7/)

December 23, 2024 at 2:35 AM
So what is the solution? There are a few different approaches I have seen. (a) you can make parametric assumptions about the relationship between the predicted and true phenotype, which can then be used to 'correct the effect estimates' as in this paper: www.biorxiv.org/content/10.1.... (7/)
Finally, we looked at a question that has often vexed me: is 'test set R2' really a good way to evaluate imputation models? Turns out the answer is no. This is because this metric is unable to disentangle variation in a phenotype due to environment from that due to genetics. (7/)

December 17, 2024 at 6:08 PM
Finally, we looked at a question that has often vexed me: is 'test set R2' really a good way to evaluate imputation models? Turns out the answer is no. This is because this metric is unable to disentangle variation in a phenotype due to environment from that due to genetics. (7/)
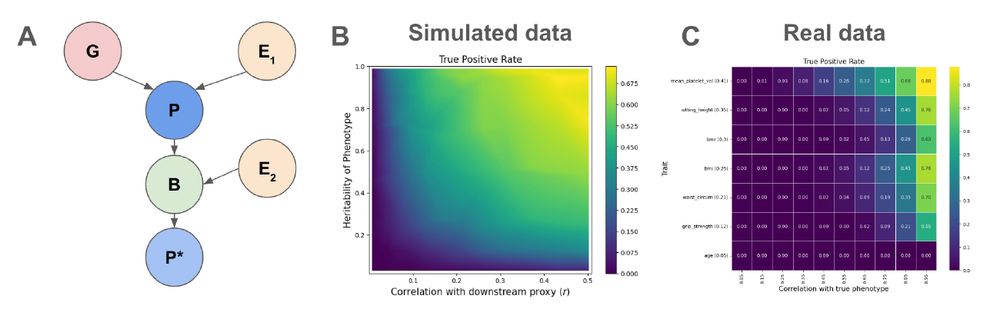
Next we looked how the heritability of phenotypes and imputation accuracy can affect power in the case of unbiased imputation using both real and simulated data. (6/)
December 17, 2024 at 6:08 PM
Next we looked how the heritability of phenotypes and imputation accuracy can affect power in the case of unbiased imputation using both real and simulated data. (6/)
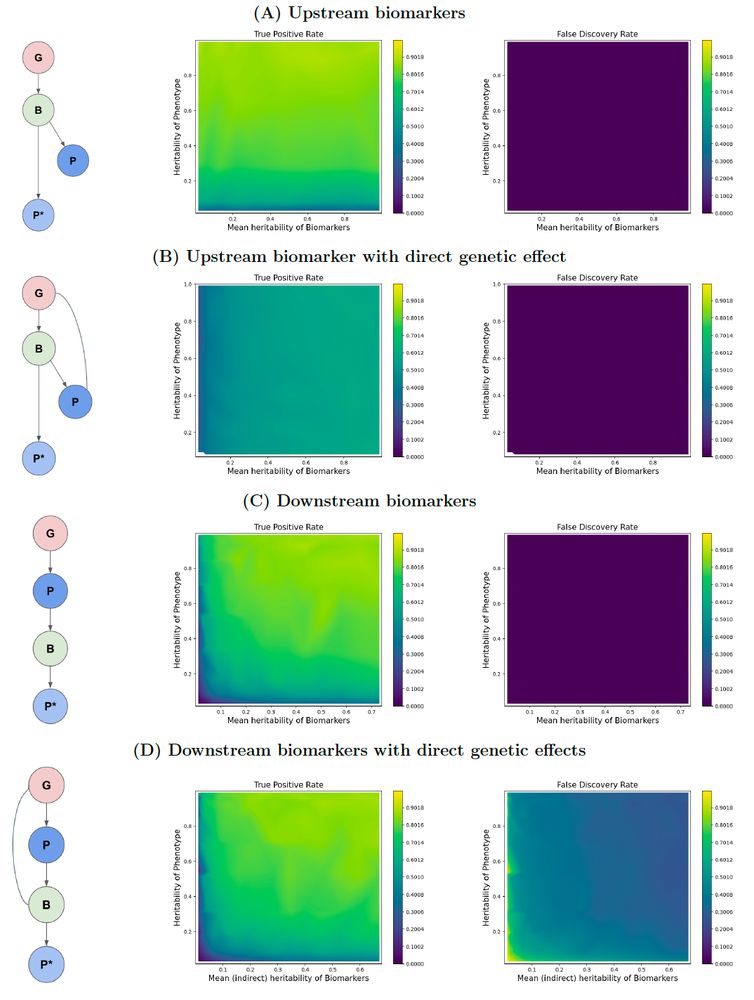
Our work is slightly orthogonal to this. We studied how the causal relationships between biomarkers used for imputation and the phenotype being imputed affects power and FDR. We started with the question 'upstream vs downstream biomarkers: which to use?' (4/)
December 17, 2024 at 6:08 PM
Our work is slightly orthogonal to this. We studied how the causal relationships between biomarkers used for imputation and the phenotype being imputed affects power and FDR. We started with the question 'upstream vs downstream biomarkers: which to use?' (4/)
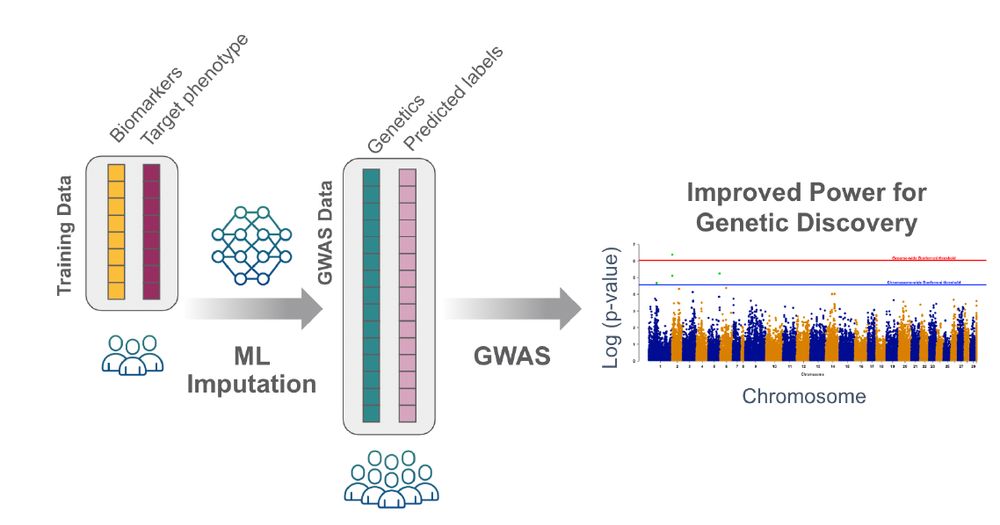
Okay friends, I am back with a new thread and this time about a new pre-print that I co-authored. Today's topic is 'ML assisted GWAS'. What is that, you ask? It's basically estimating missing values in phenotypes of interest using associated measurements (1/)
December 17, 2024 at 6:08 PM
Okay friends, I am back with a new thread and this time about a new pre-print that I co-authored. Today's topic is 'ML assisted GWAS'. What is that, you ask? It's basically estimating missing values in phenotypes of interest using associated measurements (1/)
Why is that? To understand that, let's remind ourselves how such phenotypes are derived. The following is from the preliminaries of one of my papers. (3/)

November 27, 2024 at 6:59 PM
Why is that? To understand that, let's remind ourselves how such phenotypes are derived. The following is from the preliminaries of one of my papers. (3/)
A lot of complex traits are assumed to be readouts of some upstream common factors as seen below (figure from: www.cell.com/cell-genomic.... This assumption is common among behavioral traits. In such cases, identifying these latent structures can reveal the genetic architecture of related traits.(2/

November 26, 2024 at 6:59 PM
A lot of complex traits are assumed to be readouts of some upstream common factors as seen below (figure from: www.cell.com/cell-genomic.... This assumption is common among behavioral traits. In such cases, identifying these latent structures can reveal the genetic architecture of related traits.(2/
Some example generated images. (5/)

November 18, 2024 at 4:54 AM
Some example generated images. (5/)
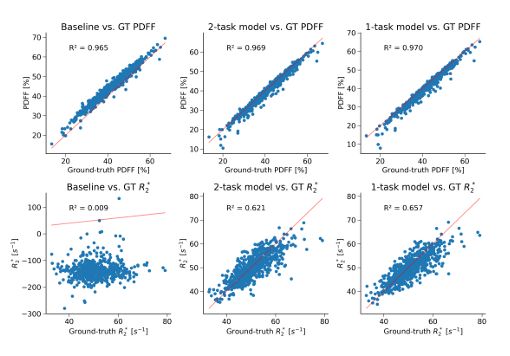
Validated on paired Dixon-IDEAL data from the UK Biobank, our approach significantly outperforms conventional baselines for R2* estimation and also provides superior estimates for PDFF. (4/)
November 18, 2024 at 4:54 AM
Validated on paired Dixon-IDEAL data from the UK Biobank, our approach significantly outperforms conventional baselines for R2* estimation and also provides superior estimates for PDFF. (4/)
By leveraging a Pix2Pix conditional GAN, our method harnesses the spatial correlation between MRI voxels to impute PDFF and R2*, thus overcoming the limitations of the Dixon MRI. (3/)

November 18, 2024 at 4:54 AM
By leveraging a Pix2Pix conditional GAN, our method harnesses the spatial correlation between MRI voxels to impute PDFF and R2*, thus overcoming the limitations of the Dixon MRI. (3/)
I presented a poster that highlighted how different causal relationships between the independent variables of such models and the outcomes can affect the power and false discovery rate of GWASs done on such 'ML-imputed' phenotypes. I am sharing the poster below for anyone who is interested. (2/)

November 18, 2024 at 4:43 AM
I presented a poster that highlighted how different causal relationships between the independent variables of such models and the outcomes can affect the power and false discovery rate of GWASs done on such 'ML-imputed' phenotypes. I am sharing the poster below for anyone who is interested. (2/)
For anyone interested in using embeddings for genetic discovery and may have missed our poster at #ashg2023. We should submit a pre-print very soon. So keep an eye out for it.

November 5, 2023 at 2:44 PM
For anyone interested in using embeddings for genetic discovery and may have missed our poster at #ashg2023. We should submit a pre-print very soon. So keep an eye out for it.