



Saez-Rodriguez Group
@saezlab.bsky.social
Account of the Saez-Rodriguez lab at EMBL-EBI and Heidelberg University. We integrate #omics data with mechanistic molecular knowledge into #opensource #ML methods
Website: https://saezlab.org/
GitHub: https://github.com/saezlab/
Website: https://saezlab.org/
GitHub: https://github.com/saezlab/
New patients can be positioned on the map:
In a cohort receiving LVADs (data from Kory Lavine’s lab), molecular changes in the map were consistent with clinical improvement, suggesting that the map may help characterize treatment responses.
In a cohort receiving LVADs (data from Kory Lavine’s lab), molecular changes in the map were consistent with clinical improvement, suggesting that the map may help characterize treatment responses.

November 6, 2025 at 12:10 PM
New patients can be positioned on the map:
In a cohort receiving LVADs (data from Kory Lavine’s lab), molecular changes in the map were consistent with clinical improvement, suggesting that the map may help characterize treatment responses.
In a cohort receiving LVADs (data from Kory Lavine’s lab), molecular changes in the map were consistent with clinical improvement, suggesting that the map may help characterize treatment responses.
This allowed us to distinguish genes driven by compositional changes from those primarily affected by molecular regulation.

November 6, 2025 at 12:10 PM
This allowed us to distinguish genes driven by compositional changes from those primarily affected by molecular regulation.
We also uncovered a network of cell-type dependencies underlying these multicellular programs, with fibroblasts playing a central role, particularly coordinating with cardiomyocyte reprogramming (where we validated several ligand candidates consistent with this interaction).

November 6, 2025 at 12:10 PM
We also uncovered a network of cell-type dependencies underlying these multicellular programs, with fibroblasts playing a central role, particularly coordinating with cardiomyocyte reprogramming (where we validated several ligand candidates consistent with this interaction).
Using Multicellular Factor Analysis, a patient-level integration method (doi.org/10.7554/eLif...), we constructed a transcriptional patient map summarizing multicellular gene-expression variation in heart failure along two main axes.

November 6, 2025 at 12:10 PM
Using Multicellular Factor Analysis, a patient-level integration method (doi.org/10.7554/eLif...), we constructed a transcriptional patient map summarizing multicellular gene-expression variation in heart failure along two main axes.
Across studies, gene-expression changes associated with heart failure showed a reproducible pattern in both bulk and single-nucleus data. Changes in cell-type composition were more variable, indicating that these two aspects of tissue remodelling may not always occur together.

November 6, 2025 at 12:10 PM
Across studies, gene-expression changes associated with heart failure showed a reproducible pattern in both bulk and single-nucleus data. Changes in cell-type composition were more variable, indicating that these two aspects of tissue remodelling may not always occur together.
We curated an extensive compendium of >1500 patients profiled w bulk or single-nuc transcriptomics, building on our previous work tinyurl.com/sn6dxu95. This data engineering effort enabled the comparison and integration of insights to generate a reference of Heart Failure.

November 6, 2025 at 12:10 PM
We curated an extensive compendium of >1500 patients profiled w bulk or single-nuc transcriptomics, building on our previous work tinyurl.com/sn6dxu95. This data engineering effort enabled the comparison and integration of insights to generate a reference of Heart Failure.
🚨 New preprint
We present an extended version of ScAPE, the method that won one of the prizes 🏆 in the @neuripsconf.bsky.social 2023 Single-Cell Perturbation Prediction challenge.
📄 preprint: doi.org/10.1101/2025...
🧬 code: github.com/scapeML/scape
We present an extended version of ScAPE, the method that won one of the prizes 🏆 in the @neuripsconf.bsky.social 2023 Single-Cell Perturbation Prediction challenge.
📄 preprint: doi.org/10.1101/2025...
🧬 code: github.com/scapeML/scape

September 19, 2025 at 7:45 AM
🚨 New preprint
We present an extended version of ScAPE, the method that won one of the prizes 🏆 in the @neuripsconf.bsky.social 2023 Single-Cell Perturbation Prediction challenge.
📄 preprint: doi.org/10.1101/2025...
🧬 code: github.com/scapeML/scape
We present an extended version of ScAPE, the method that won one of the prizes 🏆 in the @neuripsconf.bsky.social 2023 Single-Cell Perturbation Prediction challenge.
📄 preprint: doi.org/10.1101/2025...
🧬 code: github.com/scapeML/scape
🤖✨ Dive into OmniPath Explorer - Interactively browse millions of annotations & hundreds of thousands of molecular interactions. Our new AI chat agent helps you query the database effortlessly, developed by Jonathan @jonsch.one!


September 17, 2025 at 1:10 PM
🤖✨ Dive into OmniPath Explorer - Interactively browse millions of annotations & hundreds of thousands of molecular interactions. Our new AI chat agent helps you query the database effortlessly, developed by Jonathan @jonsch.one!
🥁 Check out our new preprint on OmniPath, the prior knowledge resource for #SystemsBiology, and its brand-new OmniPath Explorer web app! 🥳
📖 Preprint: www.biorxiv.org/content/10.1...
🔍 Explorer: explore.omnipathdb.org
OmniPath integrates 160+ resources for multi-omics analysis & modeling.
🧶⬇️
📖 Preprint: www.biorxiv.org/content/10.1...
🔍 Explorer: explore.omnipathdb.org
OmniPath integrates 160+ resources for multi-omics analysis & modeling.
🧶⬇️

September 17, 2025 at 1:10 PM
🥁 Check out our new preprint on OmniPath, the prior knowledge resource for #SystemsBiology, and its brand-new OmniPath Explorer web app! 🥳
📖 Preprint: www.biorxiv.org/content/10.1...
🔍 Explorer: explore.omnipathdb.org
OmniPath integrates 160+ resources for multi-omics analysis & modeling.
🧶⬇️
📖 Preprint: www.biorxiv.org/content/10.1...
🔍 Explorer: explore.omnipathdb.org
OmniPath integrates 160+ resources for multi-omics analysis & modeling.
🧶⬇️
Introducing ParTIpy, a python package for Pareto Task Inference that scales to large-scale datasets, including single-cell and spatial transcriptomics.
🔗 Manuscript: www.biorxiv.org/content/10.1...
💻 Code: partipy.readthedocs.io
🔗 Manuscript: www.biorxiv.org/content/10.1...
💻 Code: partipy.readthedocs.io

September 15, 2025 at 8:40 AM
Introducing ParTIpy, a python package for Pareto Task Inference that scales to large-scale datasets, including single-cell and spatial transcriptomics.
🔗 Manuscript: www.biorxiv.org/content/10.1...
💻 Code: partipy.readthedocs.io
🔗 Manuscript: www.biorxiv.org/content/10.1...
💻 Code: partipy.readthedocs.io
👋 Time to say goodbye to another one of our PhD students, @smuellerdott.bsky.social. She was working on the integration of phosphoproteomics and transcriptomics data and will now join the Computational Oncology Team at Merck. We wish her all the best for this new chapter! 🚀 ✨


September 1, 2025 at 5:35 AM
👋 Time to say goodbye to another one of our PhD students, @smuellerdott.bsky.social. She was working on the integration of phosphoproteomics and transcriptomics data and will now join the Computational Oncology Team at Merck. We wish her all the best for this new chapter! 🚀 ✨
🧑⚕️ Patient-level metadata analysis
- PCA + ANOVA find metabolite drivers in patient subgroups
- bioRCM spots distinct patterns; e.g., dipeptides in young vs. old kidney cancer patients
➡️ Insight into tumor microenvironment and potential biomarkers
- PCA + ANOVA find metabolite drivers in patient subgroups
- bioRCM spots distinct patterns; e.g., dipeptides in young vs. old kidney cancer patients
➡️ Insight into tumor microenvironment and potential biomarkers

August 25, 2025 at 7:07 AM
🧑⚕️ Patient-level metadata analysis
- PCA + ANOVA find metabolite drivers in patient subgroups
- bioRCM spots distinct patterns; e.g., dipeptides in young vs. old kidney cancer patients
➡️ Insight into tumor microenvironment and potential biomarkers
- PCA + ANOVA find metabolite drivers in patient subgroups
- bioRCM spots distinct patterns; e.g., dipeptides in young vs. old kidney cancer patients
➡️ Insight into tumor microenvironment and potential biomarkers
🔬 Biological Regulatory Clustering (bioRCM)
- Merges intra & exo-metabolomics (= bioRCM input)
- Groups metabolites by regulation patterns (see Figure)
🧪 Applying bioRCM, we found methionine consumption and usage within the cell, which could be linked to DNA hypermethylation in kidney cancer
- Merges intra & exo-metabolomics (= bioRCM input)
- Groups metabolites by regulation patterns (see Figure)
🧪 Applying bioRCM, we found methionine consumption and usage within the cell, which could be linked to DNA hypermethylation in kidney cancer

August 25, 2025 at 7:07 AM
🔬 Biological Regulatory Clustering (bioRCM)
- Merges intra & exo-metabolomics (= bioRCM input)
- Groups metabolites by regulation patterns (see Figure)
🧪 Applying bioRCM, we found methionine consumption and usage within the cell, which could be linked to DNA hypermethylation in kidney cancer
- Merges intra & exo-metabolomics (= bioRCM input)
- Groups metabolites by regulation patterns (see Figure)
🧪 Applying bioRCM, we found methionine consumption and usage within the cell, which could be linked to DNA hypermethylation in kidney cancer
✅ Processing, QC & Differential analysis:
- Filters low-quality features via CV & missingness
- Handles exometabolomics-specific normalization (e.g. growth rate, media blanks)
- Detects outliers
- Differential analysis (also for exometabolomics)
➡️ Ensures high-quality data
- Filters low-quality features via CV & missingness
- Handles exometabolomics-specific normalization (e.g. growth rate, media blanks)
- Detects outliers
- Differential analysis (also for exometabolomics)
➡️ Ensures high-quality data

August 25, 2025 at 7:07 AM
✅ Processing, QC & Differential analysis:
- Filters low-quality features via CV & missingness
- Handles exometabolomics-specific normalization (e.g. growth rate, media blanks)
- Detects outliers
- Differential analysis (also for exometabolomics)
➡️ Ensures high-quality data
- Filters low-quality features via CV & missingness
- Handles exometabolomics-specific normalization (e.g. growth rate, media blanks)
- Detects outliers
- Differential analysis (also for exometabolomics)
➡️ Ensures high-quality data
We present our MetaProViz #Rpackage for #metabolomics analysis & prior knowledge integration to generate mechanistic hypotheses on how metabolic changes affect metabolite classes, pathways & environment interaction
🔗
www.biorxiv.org/content/10.1...
📦
saezlab.github.io/MetaProViz/
🧵 Thread ⬇️
🔗
www.biorxiv.org/content/10.1...
📦
saezlab.github.io/MetaProViz/
🧵 Thread ⬇️

August 25, 2025 at 7:07 AM
We present our MetaProViz #Rpackage for #metabolomics analysis & prior knowledge integration to generate mechanistic hypotheses on how metabolic changes affect metabolite classes, pathways & environment interaction
🔗
www.biorxiv.org/content/10.1...
📦
saezlab.github.io/MetaProViz/
🧵 Thread ⬇️
🔗
www.biorxiv.org/content/10.1...
📦
saezlab.github.io/MetaProViz/
🧵 Thread ⬇️
✨ Key features of CORNETO:
• Unified framework for context-specific network inference
• Supports diverse omics data & network types
• Enables joint multi-sample analysis for robust, interpretable results
• Flexible: add your own constraints, hypotheses, or network models
• Unified framework for context-specific network inference
• Supports diverse omics data & network types
• Enables joint multi-sample analysis for robust, interpretable results
• Flexible: add your own constraints, hypotheses, or network models
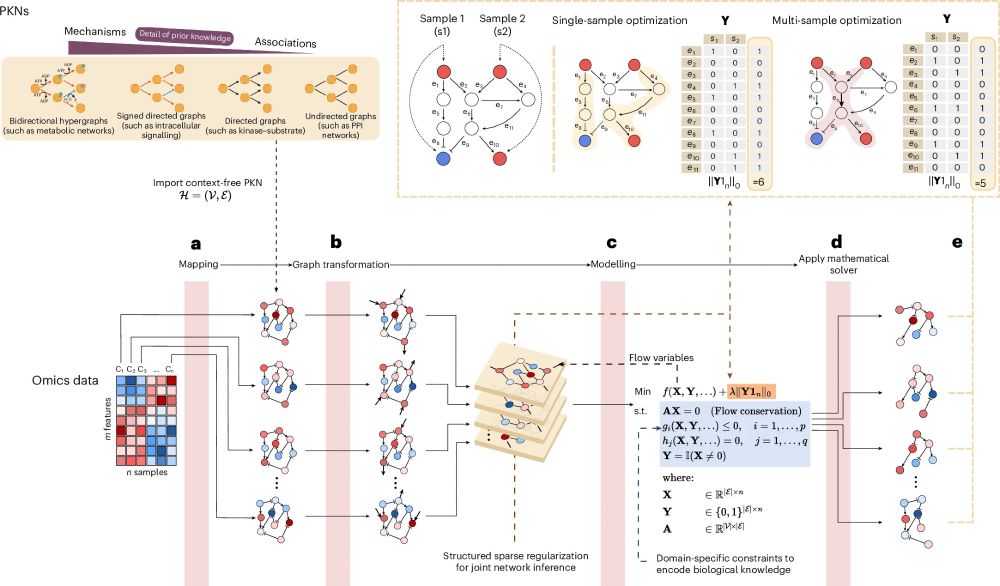
July 22, 2025 at 3:23 PM
✨ Key features of CORNETO:
• Unified framework for context-specific network inference
• Supports diverse omics data & network types
• Enables joint multi-sample analysis for robust, interpretable results
• Flexible: add your own constraints, hypotheses, or network models
• Unified framework for context-specific network inference
• Supports diverse omics data & network types
• Enables joint multi-sample analysis for robust, interpretable results
• Flexible: add your own constraints, hypotheses, or network models
🎉 The revised version of CORNETO, our unified Python framework for knowledge-driven network inference from omics data, is published in peer reviewed form
🔗 Paper: www.nature.com/articles/s42...
📖 News & Views: www.nature.com/articles/s42...
💻 Code: corneto.org
🧵 Thread 👇
🔗 Paper: www.nature.com/articles/s42...
📖 News & Views: www.nature.com/articles/s42...
💻 Code: corneto.org
🧵 Thread 👇
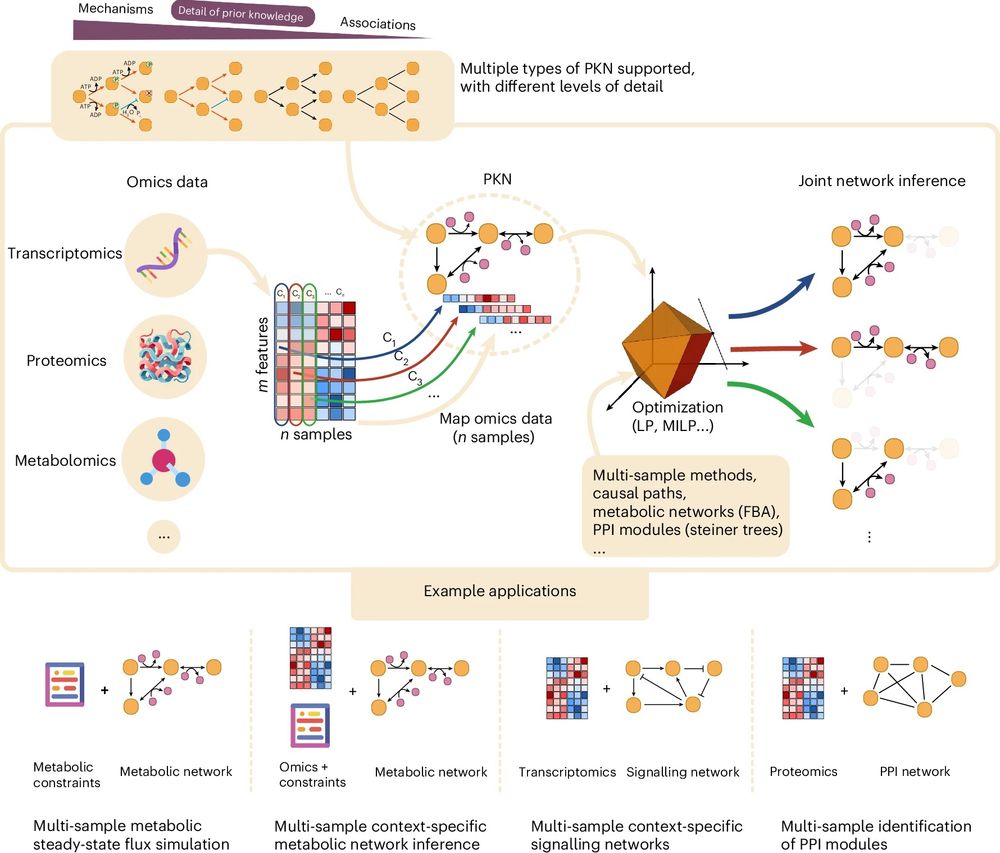
July 22, 2025 at 3:23 PM
🎉 The revised version of CORNETO, our unified Python framework for knowledge-driven network inference from omics data, is published in peer reviewed form
🔗 Paper: www.nature.com/articles/s42...
📖 News & Views: www.nature.com/articles/s42...
💻 Code: corneto.org
🧵 Thread 👇
🔗 Paper: www.nature.com/articles/s42...
📖 News & Views: www.nature.com/articles/s42...
💻 Code: corneto.org
🧵 Thread 👇
Using #siRNA knockdowns we validated these predictions and uncovered molecular processes modulating FLI1 and E2F1 transcriptional activity and their downstream effects.
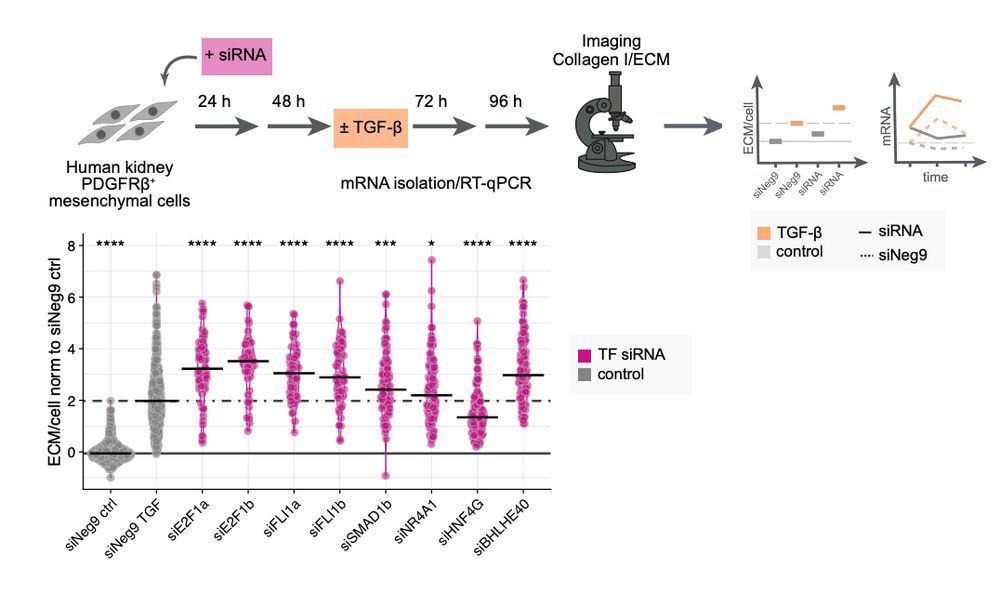
June 27, 2025 at 1:29 PM
Using #siRNA knockdowns we validated these predictions and uncovered molecular processes modulating FLI1 and E2F1 transcriptional activity and their downstream effects.
We inferred transcription factor and kinase activities using #DecoupleR tinyurl.com/decouple-R, integrated them w/ the secretomics data and generated mechanistic hypotheses related to transcriptional events modulating fibrotic progression using #COSMOS tinyurl.com/cosmos-R
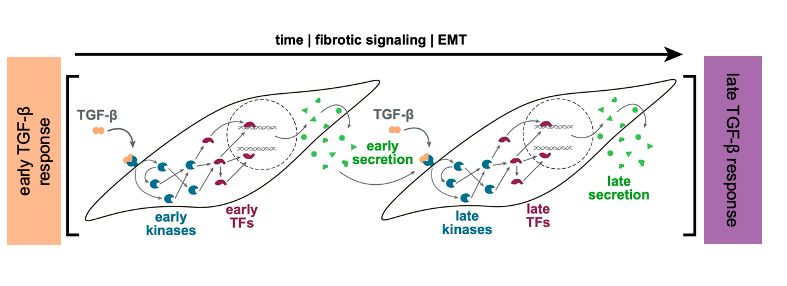
June 27, 2025 at 1:29 PM
We inferred transcription factor and kinase activities using #DecoupleR tinyurl.com/decouple-R, integrated them w/ the secretomics data and generated mechanistic hypotheses related to transcriptional events modulating fibrotic progression using #COSMOS tinyurl.com/cosmos-R
We profile fibrotic progression in response to #TGFb by quantifying COL1 deposition over time in combination with molecular data using #transcriptomics, #phosphoproteomics and #secretomics.
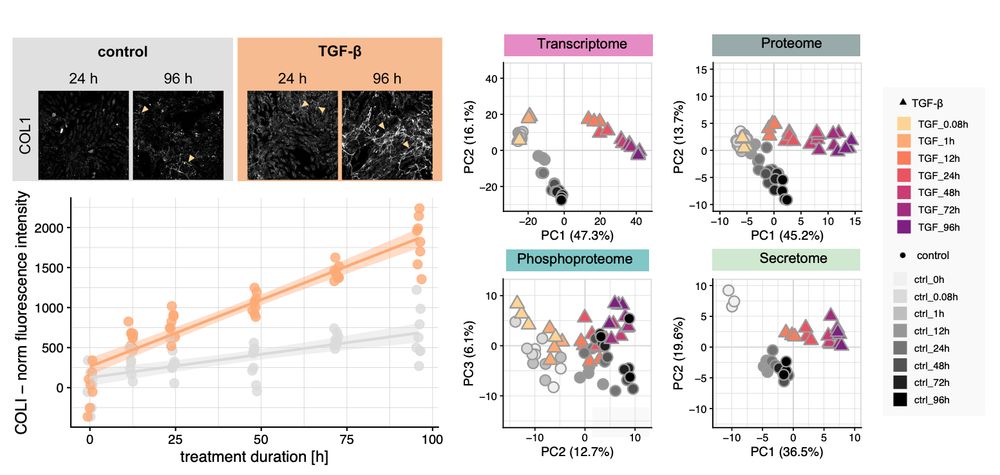
June 27, 2025 at 1:29 PM
We profile fibrotic progression in response to #TGFb by quantifying COL1 deposition over time in combination with molecular data using #transcriptomics, #phosphoproteomics and #secretomics.
The final version of our multi-omics study on kidney fibrosis is out now (tinyurl.com/kidneyfibMSB). Together w/ Pepperkok + Savitski labs @embl.org, we present a time-resolved #multiomics + computational network modeling approach in combination w/ phenotypic assays to study #kidneyfibrosis
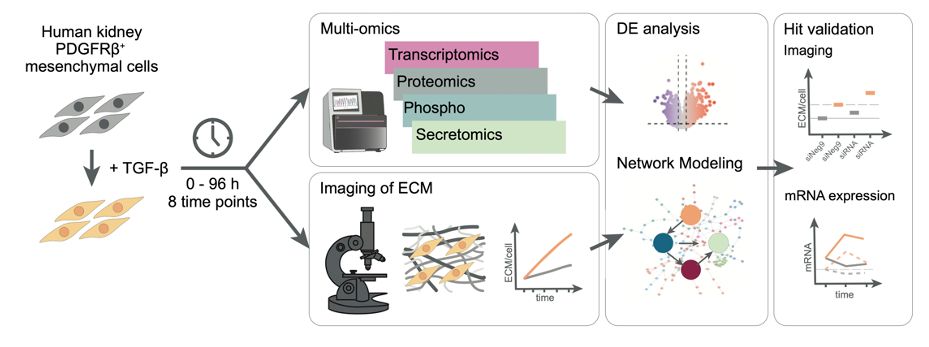
June 27, 2025 at 1:29 PM
The final version of our multi-omics study on kidney fibrosis is out now (tinyurl.com/kidneyfibMSB). Together w/ Pepperkok + Savitski labs @embl.org, we present a time-resolved #multiomics + computational network modeling approach in combination w/ phenotypic assays to study #kidneyfibrosis
👋 Wishing @paubadiam.bsky.social all the best as he leaves our lab to join the Stanford Department of Genetics in @anshulkundaje.bsky.social lab! Looking forward to his future contributions to understanding gene regulation through sequence-to-function deep learning models 💻

June 23, 2025 at 11:26 AM
👋 Wishing @paubadiam.bsky.social all the best as he leaves our lab to join the Stanford Department of Genetics in @anshulkundaje.bsky.social lab! Looking forward to his future contributions to understanding gene regulation through sequence-to-function deep learning models 💻
🔧 decoupler 2.0.6 is out thanks to @paubadiam.bsky.social, now as a core @scverse.bsky.social package scverse.org
This release includes a streamlined API aligned with scverse standards, updated vignettes, and a new one on pseudotime enrichment analysis 👇
decoupler.readthedocs.io/en/latest/
This release includes a streamlined API aligned with scverse standards, updated vignettes, and a new one on pseudotime enrichment analysis 👇
decoupler.readthedocs.io/en/latest/
June 12, 2025 at 6:36 AM
🔧 decoupler 2.0.6 is out thanks to @paubadiam.bsky.social, now as a core @scverse.bsky.social package scverse.org
This release includes a streamlined API aligned with scverse standards, updated vignettes, and a new one on pseudotime enrichment analysis 👇
decoupler.readthedocs.io/en/latest/
This release includes a streamlined API aligned with scverse standards, updated vignettes, and a new one on pseudotime enrichment analysis 👇
decoupler.readthedocs.io/en/latest/
We also introduce a tumor-based benchmark using the CPTAC data. By leveraging both proteomics & phosphoproteomics data, we identify a gold standard set of kinases with high or low activity in human tumors, which are then used to evaluate each method 🩺🧬

May 23, 2025 at 7:38 AM
We also introduce a tumor-based benchmark using the CPTAC data. By leveraging both proteomics & phosphoproteomics data, we identify a gold standard set of kinases with high or low activity in human tumors, which are then used to evaluate each method 🩺🧬
In the revised version, we significantly improved the benchmark description and included additional metrics to evaluate kinase activity inference methods 🔍. First, using perturbation experiments, we assessed how well each method is able to recover the perturbed kinases 📈🧪

May 23, 2025 at 7:38 AM
In the revised version, we significantly improved the benchmark description and included additional metrics to evaluate kinase activity inference methods 🔍. First, using perturbation experiments, we assessed how well each method is able to recover the perturbed kinases 📈🧪